Sindrome de Behçet – os desafios do diagnóstico: uma revisão sistemática
Behçet Syndrome – the challenges of diagnosis: a systematic review
Elisabete de Moura Godoy Baradelli1
Emerson Barbosa da Silva2
1Analista Clínica/Centro Universitário Saúde ABC – CUSABC. Santo André-SP, Brasil.
2Mestre em Ciências da Saúde/Centro Universitário Saúde ABC – CUSABC. Santo André-SP, Brasil.
Instituição: Centro Universitário Saúde ABC – CUSABC. Santo André-SP, Brasil.
Recebido em 14/02/2019
Aprovado em 24/03/2020
DOI: 10.21877/2448-3877.202000823
Hulûsi Behçet, um dermatologista turco, descreveu em 1937 a Doença de Behçet como “tríade clássica,” reconhecendo como sintomas, inflamações recorrentes oculares, ulcerações orais e nas regiões genitais.(1) Embora, muito provavelmente, já teria sido identificada por Hipócrates, em relatos sugestivos do início do século V a.C: “(…) muitos desenvolveram ulcerações aftosas. Muitas ulcerações sobre os genitais(…) oftalmias lacrimosas de caráter crônico, com dores; excreções fúngicas das pálpebras externamente, e internamente, que destruíram a vista de muitas pessoas (…)”. Subsequentemente, outros sintomas associados à Doença de Behçet foram sendo reconhecidos, caracterizando-a como multissistêmica, com envolvimento vascular, articular, do sistema nervoso central (SNC), pulmonar, trato gastrointestinal (TGI), cardíaco e cutâneo.(2)
A incidência das ocorrências oculares tem a variável de 23% a 96%, e, de acordo com os estudos, o envolvimento ocular constitui a primeira manifestação da doença em cerca de 10% dos casos.(3) A DB é motivo decorrente de afecções oculares graves, denominada de uveíte. Em 1979, Mishima(4) evidenciou que 12% das cegueiras contraídos em adultos jovens no Japão se deviam à DB. Uma pesquisa realizada entre 1975 e 1985 concluiu que houve redução da acuidade visual útil em 76% dos pacientes no período de seis a dez anos após início dos indícios de inflamações oculares.(5) Averiguações mais atuais propõem uma redução considerável da rapidez de agravamento da acuidade visual nos últimos dois decênios.(6)
A etiopatogenia da DB não está inteiramente esclarecida, no entanto, as informações epidemiológicas e a maior parte dos estudos a classificam como uma doença autoimune e concordam com a interação de uma agregação de fatores intrínsecos, como as predisposições genéticas,(7) e extrínsecos, como ambientais.(8) Identifica-se uma susceptibilidade elevada para a manifestação da DB nos indivíduos que exibem o alelo HLA-B51 que está presente em 50% a 77% nesses indivíduos.(7) Determinados agentes infecciosos igualmente parecem estar incluídos na patogênese desta enfermidade. A existência do DNA do vírus Herpes simplex e anticorpos agonistas do vírus foram descobertos em elevadas proporções nos enfermos com DB quando confrontados com grupos de controle. A existência de vasculite, a indispensável lesão histopatológica e imunocomplexos circulantes indicam a classificação da doença como autoimune.(9)
A Doença de Behçet é considerada uma doença rara, e se expressa em uma distribuição global com diversas prevalências, sendo mais comum na Bacia do Mediterrâneo e em países do leste europeu, conhecida historicamente como a “antiga rota da seda”. As mais elevadas taxas de predominância são achadas em Istambul, na Turquia, com cerca de quatrocentos casos por 100 mil adultos.(10) A afecção é referida em menor proporção em outros países. A prevalência estimada varia de 6,4 casos por 100 mil adultos na Espanha a 13 por 100 mil adultos no Japão.(11)
A Doença de Behçet reconhecida após como Síndrome de Behçet (SD) permanece em uma situação clínica relacionada a agravamentos, com morbimortalidades significativas. Todavia, ainda que permaneçam questões a serem respondidas, ocorre um progresso notório na compreensão de suas manifestações fisiopatológicas.
O objetivo da presente revisão literária é analisar manifestações e dados clínicos laboratoriais a fim de explorar e ampliar informações dos pacientes diagnosticados com a Síndrome de Behçet.
MATERIAL E MÉTODOS
Foi realizada uma busca e extração em bancos de dados eletrônicos PubMed, SciElo e Medline, no período de 1964 até 2012, sendo que os artigos encontrados foram traduzidos por meio do aplicativo doctranslator. Utilizamos como descritores “Behçet”, “multisystem inflammation” e o operador booleano “AND”. Como critério de inclusão foram selecionadas pesquisas com dados para diagnóstico em pacientes com a Doença de Behçet, com o intuito de explorar e ampliar os achados entre as pesquisas. Os critérios de exclusão foram: (1) trabalhos com ausências dos sintomas da Doença de Behçet, (2) trabalhos com ausência de dados para diagnóstico, (3) trabalhos repetidos e (4) trabalhos cujo tema não era o proposto para seleção. A elaboração deste estudo implicou leitura da bibliografia básica e análise das informações obtidas que passaram a fazer parte do corpo deste trabalho.
REFERENCIAL TEÓRICO
Quadro clínico
O diagnóstico é essencialmente clínico, afecções autoinflamatórias são reconhecidas por episódios idiopáticos multissistêmico, desconhecendo um antígeno ou anticorpo distintivo a retorno imune.(12)
Manifestações mucocutâneas
São os principais sintomas e mais recorrentes da SD, observadas em 97% dos casos estudados, apresentando formato oval ou redondo com bordas bem definidas. São geralmente dolorosas, rodeadas por um halo vermelho. A base necrosada é de cor esbranquiçado-amarelada. Aftoses orais localizam-se nas bochechas, lábios, gengivas, palato, amígdalas e faringe, com duração aproximada de uma a duas semanas em processo de cura espontâneo. As lesões genitais, em 65% dos casos, são semelhantes morfologicamente com as ulcerações orais, entretanto maiores, geralmente encontradas na vulva predominando nos grandes e pequenos lábios, no sexo feminino e região escrotal no sexo masculino, com o período de cura ainda mais lento, de duas a quatro semanas, com menos frequência de retorno e mais dolorosas.(13)
As lesões cutâneas se manifestam em alguns casos por pústulas, pápulas, nódulos eritematosos ou pseudofoliculite sendo esse último mais frequente, surgindo nos membros inferiores e na região púbica, podendo ser vista em todo o corpo lesões acneiformes, localizando-se preferencialmente nos membros superiores e inferiores.(14)
Manifestações oculares
As manifestações oculares abrangem cerca da metade dos pacientes, sendo que maior parte nos jovens do sexo masculino. Uveíte é a mais habitual, recorrente, bilateral e crônica, resultando em alterações estruturais, cicatrizes na retina. Quando ao hipópio (inflamação intensa), apresentando exsudado, espesso e branco, hemorragias, edema de papila e doença macular importante causa de morbidade levando a cataratas e, menos constante, a glaucoma.(15)
Manifestações articulares
Artrite, em geral não erosiva, pode se apresentar em grandes ou pequenas articulações na SD, embora os joelhos sejam os mais correntemente afetados.(14)
Manifestações do sistema nervoso central
O SB no SNC é um motivo relevante de morbilidade e mortalidade, que pode variar de 3% a 44% dos pacientes. Envolvem o parenquimatoso (80% dos casos), que compromete preferivelmente o tronco cerebral, gânglios da base, estruturas diencefálicas e cápsulas internas, provocando sintomas piramidais ou motores, mutações cognitivas, ataxia e distúrbios esfincterianos, ou trombose de seio venoso (20% dos casos) com edema de papila com ou sem cefaleia. Elevação de proteínas no LCR foram relacionadas ao pior prognóstico como meningites e compressões medulares.(16)
Manifestações vasculares
Em membros inferiores é frequente ocorrer tromboflebite e trombose venosa profunda (TVP). Oclusão ou obstrução de outros vasos calibrosos como veia cava e veias supra-hepáticas (Síndrome de Budd-Chiari) é rara. O envolvimento de vasos calibrosos, principalmente das artérias, é uma essencial causa de mortalidade. Aneurisma e/ou oclusão arterial são as preeminentes alterações encontradas. Pacientes com aneurismas de artéria pulmonar correm um maior risco de hemoptise (expectoração de sangue pelos pulmões) maciça, resultando a óbito.(14)
Manifestações gastrointestinais
Os sintomas gastrointestinais incluem anorexia, hiperêmese (vômito), náuseas, dispepsia, diarreia e enteralgia, lesões ulcerativas frequentemente nas regiões do íleo, contínuo pelo ceco e outras porções do colón, podendo surgir em outras partes do sistema digestivo.
O processo inflamatório intestinal tem como principal diagnóstico diferencial, histologicamente, as lesões que são semelhantes à Doença de Crohn; entretanto, a presença de granulomas pode ser usada para descartar SB. A cintilografia com leucócitos mostrou a presença de inflamação.(13)
Manifestações cardíacas
O envolvimento cardíaco é incomum, inclui lesões valvulares (prolapso da válvula mitral), pericardite, miocardite, trombose intracardíaca, endomiocardiofibrose, trombose/aneurisma coronarianas, angina, insuficiência cardíaca, aneurisma do ventrículo e infarto do miocárdio.(13)
Manifestações renais
É rara entre outros tipos de vasculites. Os sintomas apresentam amiloidose, doença vascular, glomerulonefrite, hematúria, proteinúria, Insuficiência Renal Aguda (IRA), mas podendo progredir para Insuficiência Renal Crônica (IRC).(15)
Manifestações pulmonares
Pode apresentar, mesmo que rara, embolia, derrame pleural, pleurisia, fibrose e infeção pulmonar.(15)
Outras manifestações
Os pacientes podem apresentar outros sintomas como febre e mal-estar. Disfunção eréctil pode se presentar em pacientes com disfunção neurológica e vascular, podendo ser observado nestes a função do trato urinário inferior.(17)
DIAGNÓSTICO CLINICO
Foi elaborado em 1990, pelo International Study Group for Behçet´s Disease (ISGBD) com a finalidade de protocolar critérios diagnósticos com um conjunto de características da SB. Apesar de tais padrões terem sido primeiramente nomeados como critérios diagnósticos, os autores enfatizam que sua utilidade é garantir a uniformidade de pacientes inclusos em estudos, assim possibilitando a comparação entre grupos, não tendo que representar como única ferramenta para diagnosticar casos individuais.(18)
Um paciente pode ser classificado como portador da síndrome de Behçet quando o critério 1 (Quadro 1) está presente, associado a quaisquer dois dos demais critérios, não necessariamente de forma simultânea.

Protocolos clínicos incluindo toda uma anamnese do paciente, identificação, história da doença, uso de medicações contínuas, antecedentes pessoais e familiares, o exame físico e a coleta de exames laboratoriais de rotina como:
Hemograma
Avaliação clínica geral e diagnóstico de processos infecciosos, leucemias/leucoses, trombocitose e trombocitopenia. O hemograma é uma das análises mais usadas na medicina, pois seus dados gerais possibilitam uma avaliação extensa da condição clínica do paciente. Embora não seja um teste extremamente sensível e específico para concluir o diagnóstico de SB, pode ser encarado como um sinal e/ou sintoma, integrante da condição inicial do paciente. No hemograma são analisadas as três séries celulares componentes do sangue: eritrócitos, leucócitos e plaquetas, constituindo o eritrograma, leucograma e plaquetograma. No eritrograma, são contados os eritrócitos, são medidas as concentrações de hemoglobina e hematócrito, são determinados os índices hematimétricos (volume celular médio, concentração de hemoglobina corpuscular média, hemoglobina corpuscular média), além da determinação do RDW, que apresentam a variação do tamanho dos eritrócitos. No leucograma, os leucócitos são contados em termos gerais, sendo classificados em uma contagem relativa em diferentes populações (neutrófilos, basófilos, eosinófilos, linfócitos, monócitos), segundo suas características citológicas. No plaquetograma, as plaquetas são contadas e seu tamanho médio e variações de volume são determinados (MPV e PDW). Todas estas análises são seguidas por microscopia após coloração para avaliação das características e/ou alterações morfológicas de cada série. Estes dados em conjunto permitem indicativos diagnósticos que, quando cruzados com outros dados e/ou resultados, são de extrema importância clínica.(20)
Velocidade de hemossedimentação (VHS)
É um exame sensível, mas pouco específico dos processos inflamatórios, indicado como marcador de atividades infecciosas inespecíficas, doenças autoimunes e reumáticas podendo elevar-se nos processos inflamatórios agudos e crônicos, nas lesões e destruição tecidual, sendo utilizado no controle progressivo das enfermidades reumáticas ou crônicas como da SB.(20)
Urina 1
É um dos exames mais usados como triagem das principais funções metabólicas do organismo, assim supervisionando a função renal dos pacientes. Hoje, a metodologia automatizada permite uma avaliação padronizada dos elementos presentes na amostra examinada por meio de leitores ópticos de tiras reagentes. Este exame visa detectar os elementos anormais e analisar o sedimento urinário em amostras de urina.(21)
Ureia
A dosagem da ureia no sangue ajuda a detectar a insuficiência renal (IR) precocemente, porém é um complemento associado a valores da creatinina, indicado para pacientes de SB que fazem uso de medicações, como, por exemplo, corticoides, que retêm líquidos, causando edema por todo o corpo. É utilizado o método cinético ultravioleta com referências em mg/dL.(20)
Creatinina
A creatinina sérica é o principal teste para a avaliação da função renal, refletindo a filtração glomerular, sendo mais sensível e específica que a ureia. É amplamente utilizada para o cálculo da estimativa de filtração glomerular pela fórmula Cockroft-Gault ou MDRD, na triagem de doença renal crônica.
- Fórmula Cockcroft-Gault: Depuração de creatinina = [(140 – idade) x peso]/creatinina sérica x 72 (x 0,85 para mulheres);
- Fórmula MDRD simplificada: RFG = 186 x creatinina sérica -1,154 x idade-0,203 x 0,742 (se mulher) x 1,212 (se afroamericano). Importante ressaltar que podem ser alterados os valores dependendo do uso de medicações, falta do repouso antes da coleta entre outros fatores.(22)
Aminotransferases
O aspartato aminotransferase (AST) e a alanina aminotransferase (ALT) são células encontradas no coração, no sistema musculoesquelético, rim e fígado e têm interesse clínico importante para o diagnóstico de lesões hepáticas e cardíacas provocadas uma vez que extravasam essas células lesadas para a corrente sanguínea.(23)
Fator antinuclear (FAN)
Por imunofluorescência indireta, o estabelecimento dos padrões do FAN é geralmente seguido pela determinação mais específica dos anticorpos contra os antígenos a eles associados. As células Hep-2 são o melhor substrato para pesquisa por fornecerem maior sensibilidade com os antígenos nucleares possíveis, indicado para diagnóstico de doenças autoimunes sistêmicas, e tem como interpretação clínica os padrões de fluorescência. Geralmente indicam o grupo de antígenos intracelulares, sugerindo a investigação posterior ou associação patológica. São considerados de importância clínica resultados superiores a 1:80. Anticorpos antinucleares, principalmente em títulos baixos, podem ser encontrados em indivíduos normais, infecções e doenças inflamatórias crônicas, devendo ser correlacionados com os dados clínicos. Algumas medicações podem estar associadas ao desenvolvimento de FAN positivo, como procainamida, hidralazina, fenotiazinas, difenilhidantoína, isoniazida, quinidina, entre outros, com títulos detectáveis por meses e até anos após a interrupção de sua administração. Tem como referência: Não reagente. Avanços técnicos no teste de FAN levaram a um aumento em sua sensibilidade e consequentemente a uma redução na sua especificidade ao utilizar diluição inicial de 1/80.(20)
Sorologia para hepatite B, C e HIV
Quando possível, compreende na detecção desses vírus e é especialmente indicada para a confirmação precoce do diagnóstico dessas afecções. Possui utilização também na resolução de resultados sorológicos indeterminados. Além disto, o método independe da resposta imune do hospedeiro, indicando a presença do vírus antes do período de soroconversão.(20)
Proteinúria 24 horas
Avaliação de doenças renais. A proteinúria não é uma doença; trata-se de um marcador clínico, indicando a existência de uma anormalidade renal evidente. Quando causada por doença renal ou sistêmica, é acompanhada por outras anormalidades clínicas tais como: creatinina sérica elevada, sedimento urinário anormal, evidência de enfermidade sistêmica (febre, erupção da pele, vasculite). Principais razões para o desenvolvimento de proteinúria: proteinúria funcional (processo benigno originado por agressores fisiológicos ou psicológicos, tais como enfermidades agudas, exercícios, estresse emocional e uma entidade bem descrita denominada “proteinúria ortostática”); superprodução de proteínas plasmáticas filtráveis circulantes (proteínas de Bence Jones, associadas com mieloma múltiplo); proteinúria glomerular (resultante de anormalidades na membrana basal glomerular); proteinúria tubular (ocorre como resultado de reabsorção defeituosa das proteínas filtradas normalmente no túbulo proximal, tendo como causas a presença de necrose tubular aguda, lesão tóxica por chumbo ou aminoglicosídeos e alterações metabólicas hereditárias, referenciando como 0,03 a 0,14g/24h.(20)
Crioglobulinas
São um grupo de proteínas que, sob baixas temperaturas, formam agregados insolúveis que se precipitam. Após aquecimento tendem a se dissolver. Indicações: Diagnóstico de croglobulinemia. Interpretação clínica:
- Crioglobulina tipo I: imunoglobulinas monoclonais, da classe IgG, IgM ou IgA (raramente proteína de Bence-Jones) sem atividade de fator reumatoide (FR) e não fixa complemento: podem estar presentes na macroglobulinemia de Waldenströn, mieloma múltiplo, púrpura, fenômeno de Raynaud.
- Crioglobulina mista do tipo II: imunoglobulinas monoclonais, classe IgM, IgG ou IgA, que geralmente têm atividade de FR, associadas a IgG policlonal e fixam complemento: são comuns em doenças infecciosas crônicas, sobretudo hepatite B e, principalmente, hepatite C, que é a principal causa de crioglobulinemia; também é observada na artrite reumatoide e síndrome de Sjögren.
- Crioglobulina mista tipo III: imunoglobulinas de origem policlonal com, pelo menos, uma com atividade de FR e que fixam complemento: doenças reumáticas como LES, artrite reumatoide, mas também ocorre em doenças infecciosas crônicas, virais, bacterianas e parasitárias.(20)
Pesquisa do HLA B51
Antígeno relacionado à doença de Behcet. Está presente em 50% a 77% dos pacientes com a doença. Indicação: Associada aos parâmetros clínicos e outros exames laboratoriais, auxilia na identificação dos indivíduos com a doença. Tem como interpretação clínica, em regiões de alta prevalência no Oriente; a detecção do antígeno está associada à presença de doença. No entanto, está presente também em indivíduos não acometidos, contudo, este resultado é analisado em função de parâmetros clínicos e outros exames laboratoriais, referenciado como não detectado.(20)
Líquor
A análise clínica inicia-se já no processo de coleta, onde deve ser verificado se o fluido corre sob pressão (indicativo de hipertensão intracraniana) ou apenas em gotejamento lento (normal). A aparência deve ser límpida e incolor. A citologia deve ser iniciada prontamente, pois as células suspensas em líquor sofrem rápida degradação in vitro. Contam-se as células presentes por milímetro cúbico com uso da câmara de Neubauer. A diluição não é necessária, a menos que seja observada celularidade muito elevada. A contagem elevada de leucócitos está associada a infecções, sendo o predomínio de neutrófilos indicativo de infecção bacteriana, e o predomínio de linfócitos associado a infecções virais e a hiperproteinorraquiana (elevação proteica) indicam meningites e compressões medulares sendo os valores de referência.(24)
Outros exames
Os autores recomendam não acrescentar como valor os resultados, porém, quando alterados, devem abrir margem às investigações adicionais, acrescidos também à atividade em sistemas orgânicos específicos, de acordo com as diferentes manifestações da SB quando há suspeita.(18) Como exames, acréscimo de exames oftalmológicos, de imagens como: Tomografia computadorizada (TC), ressonância nuclear magnética (RNM) e angiografia convencional, ultrassonografia venosa ou artérias com Doppler e/ou TC, endoscopia digestiva alta (EDA), colonoscopia e/ou exame radiológico contrastado.
Teste de patergia, que é a inserção oblíqua de uma agulha de calibre 20 Gauge na pele em condições estéreis, sem injeção de salina, deve produzir um nódulo eritematoso ou pustular no local após 24 a 48 horas para se considerar um resultado positivo.(18)
Em 2004, Lawton et al.(25) avaliaram a aplicabilidade numérica nos sintomas a fim de facilitar no diagnóstico. Foram considerados 14 ítens como medidas da atividade da SB, pelo índice denominado Behçet´s Disease Activity Index (BDAI). Neste protocolo apenas queixas e exame físico em paciente com SB, sem provas laboratoriais.

A presença dos critérios acima (Quadro 2) nas quatro semanas anteriores é utilizada para quantificar a atividade da síndrome de Behçet e um índice é produzido pela soma simples dos critérios positivos. Porém, a reprodutividade e a validade deste índice entre diferentes centros ainda não foram convenientemente avaliadas.
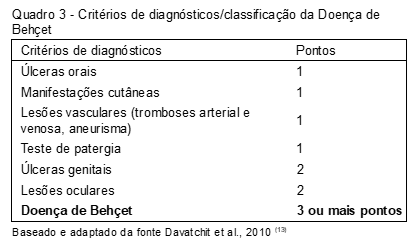
O International Criteria for Behçet´s Disease (ICBD) foi criado em 2006 para melhorar e sensibilizar o diagnóstico da SB (Quadro 3)
DISCUSSÃO
Não existem exames laboratoriais para diagnosticar a SD. Porém existem marcadores de inflamações como Proteína C Reativa (PCR), Velocidade de Hemossedimentação (VHS) e leucocitose, imunoglobulinas, fatores de complementos que podem apresentar aumentados. A associação do HLA B51 (Human Leucocyte Antigen – Antígeno Leucocitário Humano) parece apresentar uma variação geográfica, presente no Japão e população mediterrânea, mas com ausência nos EUA e Reino Unido,(26) e do MHC (Major Histocompatibility Complex – Complexo Principal de Histocompatibilidade) classe I também não diagnostica, porém pode ajudar no diagnóstico diferencial. Essa associação gerou produtos que pertencem ao grupo das “proteínas de choque térmico” (HSP – heat shock proteins), sugerindo a capacidade atuante molecular apresentando antígenos a linfócitos T. Porém, anticorpos contra o vírus herpes simples e o próprio DNA viral também foram achados em porções mais elevadas em pacientes com SD do que em controles saudáveis. Diversos agentes microbianos já ocuparam a posição de suspeitos desencadeadores da enfermidade. Algumas espécies de estreptococos achadas na flora oral já foram mencionadas como causadoras de lesões aftosas frequentes. Sorotipos incomuns de Streptococcus sanguis e anticorpos contra esta bactéria foram descobertos em flora oral e no sangue, respectivamente, nos casos de SB. Contrariamente, o conjunto de tais estudos demonstra uma condição de hiper-reatividade a antígenos comuns no meio ambiente. Entretanto, todos esses resultados não são conclusivos.(18)
Uma pesquisa no Japão observou a presença de Streptococcal Bes-1 e de DNA do Herpes simples nos tecidos de portadores de Behçet, sugerindo que infecções podem ter relevância na gênese desse quadro.(27) Albuquerque et al.(28) constatam o fato de que a apresentação da doença de forma incompleta é recorrente. Este fato pode levar a diagnósticos equivocados, exemplificando com a doença celíaca, que se acompanha de sintomas de aftas orais, diarreias frequentes, além de alterações discretas na mucosa do intestino delgado. No estudo de caso clínico presenciou-se o anticorpo antigliadina IgG reagente gero, primeiramente, uma suspeita da doença celíaca. Entretanto, não houve melhora dos sintomas com a introdução da dieta livre de glúten, o que foi determinante para a mudança de rumo da investigação.(26)
CONCLUSÃO
O diagnóstico da Síndrome de Behçet é apoiado nas manifestações clínicas associadas a complicações dos sintomas dos pacientes que devem ser analisados pelo médico. Entretanto, ainda há lacunas a serem preenchidas, como o entendimento do fator ambiental para a doença se expressar e estudos específicos para estratégias no tratamento mais eficaz. Há necessidade de profissionais de diversas áreas auxiliando no diagnóstico precoce e correto, definindo uma intervenção terapêutica eficiente a fim de se definir um prognóstico, muitas vezes alterando o rumo natural da doença e ofertando qualidade de vida ao paciente.
Abstract
Behçet’s disease (DB) is an affection understood to be multisystemic inflammation of an as yet unknown cause, presenting symptoms such as oral ulcers, genitalia, uveitis, cutaneous lesions and recurrent vasculitis. It is more frequent in countries stretching from the Mediterranean to the Far East. Currently recognized as an autoimmune disease, DB appears to aggregate carrier genetic elements and environmental triggering factors. The establishment of international criteria for a diagnosis, clinical protocols were developed to aid the activity of the disease thus allowing the standardization of research in the area, although there are no laboratory or histopathological changes defined disease, the diagnosis dependent on a careful clinical evaluation that when are determinants for the prognosis. In this article we review clinical data for the diagnosis of Behçet’s Disease.
Keywords
Behcet Syndrome; vascular diseases; autoimmune diseases
REFERÊNCIAS
- Yazici Y. From the Silk Road to the States. The Rheumatologist 7:24-26, 2010.
- Cheng TO. Some historical notes on Behçet’s disease. Chest. 2001 Feb;119(2):667-8. doi: 10.1378/chest.119.2.667-a.
- Mamo JG, Baghdassarian A. Behçet’s disease: a report of 28 cases. Arch Ophthalmol. 1964 Jan;71:4-14. doi: 10.1001/archopht. 1964.00970010020003
- Mishima S, Masuda K, Izawa Y, Mochizuki M, Namba K. The eighth Frederick H Verhoeff lecture presented by Saiichi Mishima, MD. Behçet’s disease in Japan: ophthalmologic aspects. Trans Am Ophthalmol Soc. 1979;77:225-79.
- Benezra D, Cohen E. Treatment and visual prognosis in Behçet´s disease. Br J Ophthalmol. 1986 Aug;70(8):589-92. doi: 10.1136/bjo.70.8.589.
- Mendes D, Correia M, Barbedo M, Vaio T, Mota M, Gonçalves O, Valente J. Behçet’s disease – a contemporary review. J Autoimmun. 2009 May-Jun;32(3-4):178-88. doi: 10.1016/j.jaut. 2009.02.011.
- Mizki N, Inoko H, Ohno S. Pathogenic gene responsible for the predisposition to Behçet’s disease. Int Rev Immunol. 1997;14(1):33-48. doi: 10.3109/08830189709116843.
- Ohno S. Clinical and immunological studies on ocular lesions in Behçet’s disease. In Behçet’s disease: Pathogenetic Mechanism and Clinical features. Editado por Inaba G, University Tokyo Press, Tokyo 127-136, 1982.
- Lehner T. The role of heart shock protein, microbial and autoimmune agents in the aetiology of Behçet’s disease. Int Rev Immunol. 1997;14(1):21-32. doi: 10.3109/08830189709116842.
- Azizlerli G, Köse AA, Sarica R, Gül A, Tutkun IT, Kulaç M, et al. Prevalence of Behçet’s disease in Istambul, Turkey . Int J Dermatol. 2003 Oct;42(10):803-6. doi: 10.1046/j.1365-4362.2003. 01893.x.
- Davatchi F, Shahram F, Chams C, Chams H, Nadji A. Behçet’s disease. Acta Medica Iranica 43(4):233-242, 2005.
- Owlia MB, Mehrpoor G. Behcet’s Disease: New Concepts in Cardiovascular Involvements and Future Direction for Treatment. ISRN Pharmacol. 2012;2012:760484. doi: 10.5402/2012/760484.
- Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, et al. Behcet’s disease: from East to West. Clin Rheumatol. 2010 Aug;29(8):823-33. doi: 10.1007/s10067-010-1430-6.
- Sachetto, Z. Doença de Behçet: dados demográficos e manifestações clínicas em 87 pacientes acompanhados no ambulatório de vasculites do Hospital das Clínicas da Cidade de Campinas. Tese de Doutorado, 2011. Disponível em: <http://www.repositorio. unicamp.br/handle/REPOSIP/310636>
- Saadoun D; Wechsler B. Behçet’s disease. Orphanet J Rare Dis. 2012 Apr 12;7:20. doi: 10.1186/1750-1172-7-20
- Yurdakul S, Yazici H. Behçet’s syndrome. Best Pract Res Clin Rheumatol. 2008 Oct;22(5):793-809. doi: 10.1016/j.berh. 2008. 08.005.
- Erdogru T, Koçak T, Serdaroglu P, Kadioglu A, Tellaloglu S. Evaluation and therapeutic approaches of voiding and erectile dysfunction in neurological Behçet’s syndrome. J Urol. 1999 Jul;162(1):147-53. doi: 10.1097/00005392-199907000-00036.
- Neves FS, Moraes JCB, Gonçalves CR. Síndrome de Behçet: à procura de evidências. Rev. Bras. Reumatol. [online]. 2006, vol.46, suppl.1, pp.21-29. ISSN 1809-4570. https://doi.org/10.1590/S0482-50042006000700005.
- Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990 May 5;335 (8697):1078-80.
- Detalhes de exames. Laboratório Alvaro de apoio, São Paulo, 2018. Disponível em: <https://alvaroapoio.com.br/exames>. Acesso em: 20, agosto e 2018.
- Birch DF, Fairley KF, Becker GJ, Kincaid-Smith P. Microscopia Urinária – texto & Atlas. São Paulo, SP: Editorial Premier, Henry, JB,2001.
- Burton RD. Diagnosis of acute tubular necrosis and prerenal disease. In. Burton DR. Up to date, version 13.2, 2005. Kirsztajn GM. Avaliação do ritmo de filtração glomerular. J Bras Patol Med Lab; 43(4): 257-64 2007.
- Pinto SB. Comparação entre as dosagens de AST (Aspartato Aminotransferase) e ALT (Alanina Aminotransferase) em presença e na ausência de piridoxal fosfato). Porto Alegre, 2010.
- Strasinger, Susan K. Uroanálise e Fluidos Biológicos. São Paulo, SP: Editorial Médica Panamericana, 2aedição, ISBN 85-303-0019-X, 1991.
- Lawton G, Bhakta BB, Chamberlain MA, Tennant A. The Behcet’s disease activity index. Rheumatology (Oxford). 2004 Jan;43(1):73-8. doi: 10.1093/rheumatology/keg453.
- Pires ALG, Picarelli MMC. Doença de Behçet na adolescência: relato de um caso com boa resposta à sulfasalazina. Rev. AMRIGS, Porto Alegre, 48 (2): 104-108, abr.-jun. 2004.
- Fresco I. Highlights of the 10th International Congress on Behçet disease. Clin Exp Rheumatol. 2002 Jul-Aug;20(4 Suppl 26):S59-64.
- de Albuquerque PR, Terreri MT, Len CA, Hilário MO. Doença de Behçet na infância [Behçet’s disease in childhood]. J Pediatr (Rio J). 2002 Mar-Apr;78(2):128-32. Portuguese. doi: 10.2223/jped.827.
Correspondência
Elisabete de Moura Godoy Baradelli
Av. Lauro Gomes, 2000 – Vila Sacadura Cabral
09060-870 – Santo André- SP, Brasil