Leucemia Mieloide Crônica: aspectos clínicos, diagnóstico e principais alterações observadas no hemograma
Chronic Myeloid Leukemia: clinical aspects, diagnosis and main changes observed in complete blood count
Fernanda Roberta Sossela1
Barbara Catarina de Antoni Zoppas2
Liliana Portal Weber3
1Farmacêutica Bioquímica – Setor de Hematologia, Hospital Geral. Fundação Universidade de Caxias do Sul. Caxias do Sul, RS, Brasil.
2Professora de Parasitologia e Micologia da Universidade de Caxias do Sul. Caxias do Sul, RS, Brasil.
3Professora de Imunologia e Hematologia da Universidade de Caxias do Sul. Caxias do Sul, RS, Brasil.
Instituição: Universidade de Caxias do Sul. Caxias do Sul, RS, Brasil.
Artigo recebido em 23/10/2016
Artigo aprovado em 24/01/2017
DOI: 10.21877/2448-3877.201700543
Resumo
A Leucemia Mieloide Crônica (LMC) é uma neoplasia hematológica que corresponde de 15% a 20% de todas as leucemias. Na maioria dos casos, há expressão do cromossomo Filadélfia e a produção de uma oncoproteína com atividade tirosina-quinase aumentada. O curso clínico da doença é caracterizado por três fases: crônica, acelerada e crise blástica. O diagnóstico é estabelecido por aspectos clínicos e hematológicos. Entre os principais métodos diagnósticos pode-se citar o hemograma, o mielograma e as análises citogenéticas e moleculares. Em meio às metodologias cada vez mais sensíveis e específicas, o hemograma se constitui numa ferramenta de elevada importância como método de triagem para novos casos, principalmente quando não há sintomatologia característica. Algumas alterações típicas podem ser evidenciadas no hemograma, como o aumento significativo na contagem de leucócitos, acompanhado do aumento de basófilos e do aparecimento de células imaturas. O tratamento de primeira escolha indicado atualmente é o quimioterápico mesilato de imatinibe, que vêm apresentando sucesso terapêutico, acarretando na diminuição do número de casos de evolução da fase crônica para a fase acelerada. Neste artigo, serão discutidos os principais aspectos clínicos da doença e métodos diagnósticos, com enfoque nas alterações características encontradas no hemograma.
Palavras-chave
Cromossomo Filadélfia; Leucemia mieloide de fase crônica; Contagem de células sanguíneas
INTRODUÇÃO
A Leucemia Mieloide Crônica (LMC) é uma neoplasia hematológica, caracterizada como uma doença proliferativa do sistema hematopoiético na qual ocorre expansão clonal de uma célula-tronco pluripotente ou stem cell.(1-3) Esta célula possui a capacidade de se diferenciar em células das linhagens mieloide, linfocítica, monocítica ou megacariocítica.(2)
A LMC corresponde a aproximadamente 15% a 20% de todas as leucemias, com incidência de um a dois casos em cada 100 mil indivíduos.(3-6) Apresenta maior frequência em adultos, com faixa etária entre 40 e 60 anos, principalmente do sexo masculino. Entretanto, pode acometer indivíduos de todas as faixas etárias, com menos de 10% dos casos de pacientes com até 20 anos.(3-8)
Em aproximadamente 90% a 95% dos casos, há expressão do cromossomo Filadélfia, produto de uma translocação recíproca dos braços longos dos cromossomos 9 e 22, dando origem ao gene quimérico BCR-ABL.(1-3) Nesta translocação, há a associação do gene c-ABL (Abelson murine leukemia) no cromossomo 9 com uma porção do gene BCR (breakpoint cluster region) do cromossomo 22, t(9;22)(q34;q11), gerando a expressão e consequente tradução de uma oncoproteína com atividade tirosina-quinase aumentada, p210BCR-ABL, que é característica dos pacientes com LMC.(1-4,6,8,9) A hiperatividade da p210BCR-ABL é responsável pela oncogênese inicial da LMC, favorecendo a liberação de efetores de proliferação celular e inibidores de apoptose da célula progenitora hematopoiética.(3,6,9)
O principal fator de risco para o desenvolvimento da LMC é a exposição à radioatividade, que também é fator de risco para o surgimento de outros tipos de câncer.(8) A maior incidência de LMC pode ser observada em pacientes submetidos à radioterapia, bem como sobreviventes das regiões atingidas pelas bombas atômicas durante a Segunda Guerra Mundial. No entanto, mesmo com essa provável relação entre LMC e radiação ionizante, na maioria dos casos não há fator predisponente conhecido.(6)
O diagnóstico da LMC pode ser estabelecido por meio de aspectos clínicos e hematológicos. Apesar do surgimento de técnicas cada vez mais sensíveis e específicas na identificação citogenética e molecular do cromossomo Filadélfia, do gene BCR-ABL e seus produtos, a utilização do hemograma como exame de triagem para novos casos torna-se essencial. Muitos pacientes não apresentam sintomas específicos, dessa forma o hemograma pode sinalizar alterações características da LMC e auxiliar no diagnóstico clínico.
Este artigo constitui uma revisão que objetiva abordar as principais características clínicas da LMC, sintomatologia, formas de tratamento e diagnóstico, com enfoque nas alterações observadas no hemograma, no intuito de auxiliar os profissionais na sua utilização como método de triagem e diagnóstico para a LMC.
CURSO CLÍNICO E SINTOMATOLOGIA
A maior parte dos pacientes apresenta uma fase crônica, que dura geralmente de três a cinco anos em tratamento com fármacos convencionais, com aparecimento de poucos sinais e sintomas; uma fase acelerada, com aparecimento de blastos no sangue periférico e/ou na medula óssea; e uma fase aguda (crise ou transformação blástica), com duração de três a seis meses.(7,8) Em alguns casos, a doença pode evoluir da fase crônica diretamente para a fase de crise blástica.(3) A evolução da fase crônica para outros estágios da doença pode ser uma consequência da instabilidade genética e evolução clonal, com anormalidades cromossômicas adicionais provindas da proliferação celular induzida pelo gene BCR-ABL.(6)
O quadro clínico apresenta heterogeneidade de sintomas em todas as fases da LMC.(3) Entre as manifestações clínicas na fase inicial da doença pode-se citar anemia, artralgia, parestesia palmar, hepatoesplenomegalia, além de sintomas de hipercatabolismo, como fadiga, perda ponderal, sudorese noturna e febre.(3,6-8) A ocorrência de hemorragia e complicações trombóticas pode ser observada em menos de 5% dos casos na fase crônica.(6)
Os sinais clínicos relatados na fase de crise blástica incluem palidez, aumento da hepatoesplenomegalia, equimoses fáceis e refratariedade ao tratamento, até então eficaz na fase crônica.(7) Pode-se citar ainda a ocorrência de sangramentos, falência de múltiplos órgão e infecções, com sobrevida de três a seis meses para pacientes sem tratamento.(6)
DIAGNÓSTICO
Geralmente, o diagnóstico torna-se evidente pelos aspectos clínicos e hematológicos.(7) Podem ser utilizados os seguintes métodos para o estabelecimento de diagnóstico de LMC: medição da esplenomegalia, realização de hemograma completo, mielograma, biópsia de medula com coloração pela prata (para avaliar a presença de fibrose), cariótipo da medula óssea, PCR-qualitativo (para identificação do transcrito BCR-ABL) e PCR quantitativo. A partir dos resultados obtidos, é possível definir a fase de evolução da doença.(4)
A maior parte dos pacientes já apresenta sintomas ao diagnóstico, com a doença já estabelecida. No entanto, um número cada vez maior de pacientes está sendo diagnosticado em exames periódicos através do hemograma, estando ainda assintomáticos.(7) Desse modo, o hemograma apresenta-se como uma ferramenta importante para a identificação da LMC, e sua correta interpretação torna-se essencial para o direcionamento do diagnóstico. Neste âmbito, alguns aspectos principais devem ser observados e estão representados na Tabela I.
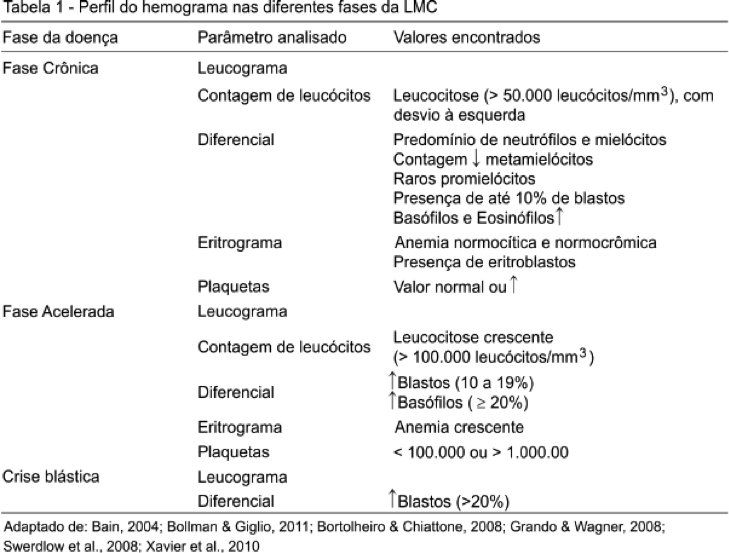
Em estágios iniciais da doença, os primeiros aspectos observados no sangue periférico são o aumento de basófilos, trombocitose e um baixo escore de fosfatase alcalina leucocitária (LAP), teste que vem perdendo a força devido ao surgimento das análises citogenéticas e moleculares.(7,11) Após, ocorre o aumento do total de leucócitos, de neutrófilos e começam a surgir células imaturas. Em alguns pacientes, há alterações cíclicas na contagem de leucócitos em intervalos de 50 a 70 dias, com uma fase de níveis leucêmicos e uma fase de níveis normais. Nas fases iniciais, há necessidade de estabelecer diagnóstico diferencial entre LMC, neutrofilia reacional e outros tipos de leucemias mieloides.(7)
Com a evolução da doença, um dos principais critérios de identificação da crise blástica é a contagem de blastos. Segundo a Organização Mundial da Saúde (OMS), o número de blastos deve ser igual ou superior a 20%.(10) Outra classificação bastante usada em estudos terapêuticos para LMC, do grupo M.D. Anderson Cancer Center (MDACC), propõe que a contagem de blastos deve ser igual ou superior a 30% ou, ainda, uma soma de blastos e promielócitos igual ou superior a 30%.(12) Conforme o estudo de Cortes et al.,(12) indivíduos com contagens de blastos de 20% a 29% apresentam sobrevida e resposta citogenética melhores do que pacientes com contagens acima de 30%, o que demonstra que o ponto de corte de blastos ainda permanece em constante discussão.
Outra questão importante é a identificação da natureza da população blástica, que pode ser realizada por meio da imunofenotipagem blástica. Na maioria dos casos, a população é de linhagem mieloide (podendo ser mieloblástica ou mieloblástica/megacarioblástica) ou mista (linfoblástica/ mieloide).(7) Em aproximadamente 70% dos casos, a transformação blástica é mieloide, podendo acontecer em múltiplas linhagens ou predominar em uma delas: mieloblástica, basofílica, eosinofílica, megacarioblástica, monocítica ou eritroblástica. A transformação linfoide ocorre em 20% a 30% dos pacientes e a transformação bifenotípica é mais raramente encontrada.(3,6)
No âmbito medular, o diagnóstico pode ser realizado por meio do mielograma e biópsia de medula. A medula óssea dos pacientes com LMC é hipercelular, em função da hiperplasia mieloide, apresentando células em todas as fases de maturação, e a série eritroide encontra-se diminuída. Pode ser observada ainda proliferação de megacariócitos, diferentes graus de fibrose reticulínica e vascularização.(3,11)
Para confirmação diagnóstica e monitoração do tratamento, são utilizadas as análises citogenéticas e moleculares.(2,7,11) Na maior parte dos casos de LMC, há presença do cromossomo Filadélfia.(7) A presença do cromossomo ou de seu transcrito BCR-ABL determina o diagnóstico da doença.(11) Na monitoração da resposta citogenética, podem-se citar as técnicas de cariótipo medular e hibridização in situ por fluorescência (FISH).(4,13) Através do cariótipo medular pode-se quantificar o número de metáfases Ph+ e detectar anomalias cromossômicas adicionais no momento do diagnóstico e a cada seis meses, até a resposta citogenética completa, e, após, a cada um a dois anos, para a identificação de eventuais recaídas.(4) O método de FISH é útil na demonstração de translocação 9-22 e detecção do gene BCR-ABL, podendo contribuir na identificação de doença residual mínima (DRM), mas não detecta as demais anomalias cromossômicas e não possui valor prognóstico estabelecido.(4,13)
A análise molecular com a utilização do método de Transcricão Reversa e Reação em Cadeia da Polimerase quantitativo, RT-qPCR (Reverse Transcription – quantitative Polymerase Chain Reaction), apresenta alta correlação com os resultados obtidos pela análise citogenética.(2) Por meio deste método, é possível identificar o rearranjo BCR/ABL e determinar o número de cópias de mRNA produzido pela p210BCR/ABL, sendo muito útil na detecção de doença residual após quimioterapia ou transplante de medula óssea.(2,4,9) A monitoração de resposta molecular deve ser realizada no intervalo de três a seis meses até a resposta molecular completa e, depois, a cada seis meses.(4)
TRATAMENTO
No tratamento da LMC, pode ser utilizada a terapia celular por meio do transplante de células-tronco hematopoiéticas, ou o tratamento medicamentoso, com a utilização de bussulfan, hidroxiureia, interferon alfa ou inibidores da tirosina-quinase.(8,9) Atualmente, o mesilato de imatinibe, inibidor da tirosina-quinase, é considerado tratamento de primeira linha para estes pacientes.(4,8,9) O imatinibe age como inibidor específico da proteína BCR-ABL, por meio da competição pelo sítio de ligação de ATP da tirosina-quinase, bloqueando a fosforilação de substratos relacionados com a regulação do ciclo celular, reativando o mecanismo morte celular.(6,9) No entanto, pode haver o desenvolvimento de resistência ao fármaco, através de mutações na região do sítio catalítico da proteína no gene BCR-ABL, incapacidade de manutenção de concentrações adequadas de fármaco no interior da célula e/ou duplicação do cromossomo Filadélfia.(8) Com o sucesso cada vez maior dos inibidores da tirosina-quinase, o número de casos de evolução da fase crônica para a fase acelerada vem diminuindo de forma expressiva.(3,9) Mesmo com o uso de imatinibe, a sobrevida dos pacientes na fase acelerada pode ser estimada em um a dois anos.(3)
O uso de mesilato de imatinibe geralmente é bem tolerado pelos pacientes, entretanto a principal preocupação é o desenvolvimento, a longo prazo, de uma segunda neoplasia, que pode ser observada também com o uso de outros fármacos utilizados no tratamento da LMC.(8)
Atualmente, estão sendo utilizados no tratamento também os inibidores da tirosina-quinase de segunda geração, como o dasatinibe, o nilotinibe e o bosutinibe, que demonstram eficácia na maioria dos pacientes resistentes ou intolerantes ao imatinibe.(4,6)
Na avaliação do sucesso terapêutico, a resposta hematológica completa é definida pelos seguintes critérios: plaquetas £ 450 mil, leucócitos £ 10.000, com diferencial normal, basófilos menor que 5% e ausência de esplenomegalia.(6) O hemograma deve ser repetido a cada duas semanas até que a resposta hematológica completa seja obtida.(4)
A terapêutica curativa para pacientes com LMC é o alotransplante de células progenitoras hematopoiéticas. Entretanto, não é considerado como terapêutica de primeira linha, sendo mais indicado para pacientes jovens e nos casos em que ocorre resposta insatisfatória ou resistência aos inibidores da tirosina-quinase.(4)
CONCLUSÃO
A LMC consiste numa neoplasia de sintomatologia muito diversa e com casos assintomáticos frequentes, de modo que a avaliação laboratorial do paciente torna-se essencial. As técnicas utilizadas demonstram-se cada vez mais avançadas, possibilitando análises citogenéticas e moleculares precisas, que permitem não somente a confirmação do diagnóstico como também a monitoração da evolução da doença e da terapêutica. No entanto, o hemograma ainda apresenta grande relevância por ser uma metodologia de fácil acesso e, quando realizado por profissionais treinados, pode auxiliar na identificação de novos casos e no acompanhamento da LMC.
Quanto às formas de tratamento, o mesilato de imatinibe continua sendo a terapia de primeira escolha. Nos casos de resistência ao imatinibe, surgem como alternativa os inibidores da tirosina-quinase de segunda geração.
Abstract
Chronic myeloid leukemia (CML) is an hematological tumor, corresponding 15 to 20% of all leukemias. In most cases, there expression of the Philadelphia chromosome and production of an oncoprotein with enhanced tyrosine-kinase activity. The clinical course of the disease is characterized by three phases: chronic, accelerated and blast crisis. The diagnosis is established by clinical and hematological aspects. Among the main diagnostic methods, there is the complete blood count, the bone marrow examination and cytogenetic and molecular analyzes. Amid the increasingly sensitive and specific methodologies, the complete blood countis an important tool as a screening method for new cases, especially when there are no characteristic symptoms. Some typical changes can be highlighted in the complete blood countis as a significant increase in leukocyte count, accompanied by increase in basophil count and the appearance of immature cells. The treatment of choice is currently the quimioterapic imatinib mesylate, which have shown therapeutic success, resulting in decrease in the number of cases of evolution of chronic phase to accelerated phase. In this article, the main clinical aspects of the disease and diagnostic methods will be discussed, focusing on characteristic changes found in the blood count
Keywords
Philadelphia Chromosome; Leukemia, Myeloid, chronic-phase; Blood cell count
REFERÊNCIAS
- Andrade GV. Papel da p190BCR-ABL como parâmetro de recaída na leucemia mieloide crônica. Rev Bras Hematol Hemoter. 2008; 30(4):297-302.
- Barboza LP, Souza JM, Simões FV, Bragança IC, Abdelhay E. Análise dos transcritos da translocação t(9;22) em Leucemia Mieloide Crônica. Rev Bras Hematol Hemoter. 2000;22(2):89-98.
- Bortolheiro Tc, Chiattone CS. Leucemia Mieloide Crônica: história natural e classificação. Rev Bras Hematol Hemoter.. 2008; 30 (supl. 1): 3-7.
- Almeida A, Castro I, Coutinho J, Guerra L, Marques H, Pereira AM. Recomendações para o diagnóstico, tratamento e monitorização da Leucemia Mieloide Crónica. Acta Med Port. 2009;22:537-44.
- Bergantini Ap, Castro Fa, Souza Am, Fett-Conte AC. Leucemia mieloide crônica e o sistema Fas-FasL. Rev Bras Hematol Hemoter.. 2005;27(2):120-25.
- Bollmann Pw, Giglio A. Leucemia mieloide crônica: passado, presente, futuro. Einstein. 2011;9(2):236-43.
- Bain BJ. Células Sanguíneas: Um Guia Prático. 4ª ed. Porto Alegre: Artmed; 2007. 488 p.
- Castro Ma, Castro Ma, Peleja Sb, Barbosa Ap, Tavares Ap, Roberti MR. Ocorrência de Múltiplas Neoplasias em Paciente Portador de Leucemia Mieloide Crônica: Relato de Caso. Revista Brasileira de Cancerologia. 2012; 58(2): 251-255.
- Grando Ac, Wagner S. Avaliação laboratorial da doença residual mínima na leucemia mieloide crônica por Real-Time PCR. J Bras Patol Med Lab. 2008; 44 (6): 433-440.
- Swerdlow Sh, Campo E, Harris Nl, Jaffe Es, Pileri Sa, Stein H, Thiele J, Vardiman Jw. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008.
- Xavier Rm, Dora Jm, Souza Cfm, Barros E. Laboratório na prática clínica: consulta rápida. Porto Alegre: Artmed, 2010.928 p.
- Cortes Je, Talpaz M, O’brien S, Faderi S, Garcia-Manero G, Ferrajoli A. Staging of chronic myeloid leukemia in the imatinib era. An evaluation of the World Health Organization proposal. Cancer. 2006;106(6):1306-15.
- Naoum PC. Avanços tecnológicos em hematologia laboratorial. Rev Bras Hematol Hemoter. 2001;23(2):15-23.
Correspondência
Fernanda Roberta Sossela
Rua Francisco Getúlio Vargas, 1130
95070-560 – Caxias do Sul, RS