A interação entre Hb B2, Hb S e beta talassemia: um relato de caso familiar
The interaction between Hb B2, Hb S and beta thalassemia: a family case report
Raphael de Souza Santos1
João Vinícius Moraes Costa2
Luiz Arthur Calheiros Leite3
Claudia R. Bonini-Domingos4
1 Aluno do Curso de Ciências Biológicas e Bolsista de Iniciação Científica PIBIC/CNPq do Instituto de Biociências, Letras e Ciências Exatas, Laboratório de Hemoglobinas e Genética das Doenças Hematológicas (LHGDH), UNESP – São José do Rio Preto, São Paulo, Brasil.
2 Aluno do curso de Biomedicina do Centro Universitário CESMAC, Maceió, Alagoas, Brasil.
3 Doutor em Bioquímica e Fisiologia, Universidade Federal de Pernambuco, UFPE e professor da pós-graduação lato sensu em Hematologia e Hemoterapia do Centro Universitário Cesmac, Maceió, Alagoas, Brasil.
4 Professora Doutora Departamento de Biologia, Coordenadora do Laboratório de Hemoglobinas e Genética das Doenças Hematológicas (LHGDH), UNESP – São José do Rio Preto, São Paulo, Brasil.
Recebido em 11/05/2021
Aprovado em 02/03/2022
DOI: 10.21877/2448-3877.202202142
INTRODUÇÃO
As hemoglobinopatias são um grupo heterogêneo de doenças genéticas hereditárias caracterizadas por defeitos qualitativos na molécula de hemoglobina (Hb), resultando em variantes por mutações pontuais ou por anormalidades quantitativas nos genes reguladores para a síntese das cadeias polipeptídicas da hemoglobina, como as doenças falciformes e as talassemias, respectivamente.(1)
A Hb S é o resultado de uma mutação pontual que leva à substituição de um resíduo de aminoácido na posição seis da subunidade da beta globina (β6Glu-Val) e que, quando herdada em homozigose (Hb SS), causa anemia falciforme. A interação entre Hb S e beta talassemia é uma condição comum decorrente da co-herança de um alelo mutante da hemoglobina S (Hb S) e de outro que carrega um dos alelos mutantes da beta talassemia, sendo representado por Hb S/β tal. Os portadores desta doença falciforme apresentam quadro clínico que varia de anemia levemente sintomática a grave, decorrente da grande variedade de alelos da beta talassemia mutantes, parcialmente (β+) ou totalmente (β0) inativos para a produção da cadeia de beta globina.(2,3)
Em termos de diagnóstico laboratorial, diferenciar Hb S/β0 de Hb S/β+ apenas pelos valores hematológicos pode apresentar alguma dificuldade, resultante da variabilidade clínica individual. Exames laboratoriais específicos e teste de biologia molecular, com estudo familiar, são necessários para estabelecer heranças e definir o perfil de hemoglobina do paciente.(4)
A hemoglobina B2 (Hb B2) é uma variante decorrente da mutação no gene da delta globina, causando a troca de um aminoácido Glicina (Gly) por uma arginina (Arg) na 16ª posição da cadeia delta globina.(3,4) Em portadores da variante Hb B2, os valores de hemoglobina A2 (Hb A2) são reduzidos em razão da síntese de cadeias delta globina mutantes em heterozigose. Além disso, os indivíduos portadores de Hb B2 não apresentam manifestações clínicas significativas, devido ao baixo nível de produção dessa variante e ao conteúdo total de Hb nos eritrócitos.(5)
O objetivo do presente estudo foi descrever um caso de interação entre Hb S/β tal e Hb B2 em um núcleo familiar de uma criança que teve o diagnóstico primário de anemia falciforme realizado por meio de testes de triagem neonatal.
RELATO DE CASO
Criança do sexo masculino, 1 ano e 9 meses, miscigenada, acompanhada em centro de referência em triagem neonatal, após diagnóstico prévio de anemia falciforme pelo Programa de Triagem Neonatal no Brasil. Clinicamente, a criança apresentava crises dolorosas, infecções recorrentes desde o nascimento e recebia transfusões de hemácias esporadicamente. Diante da constatação de divergência entre o quadro clínico apresentado pela criança e o esperado para a anemia falciforme, foi solicitado estudo genético da família (pai, mãe e filho), para elucidar o diagnóstico inicial. As amostras de sangue foram submetidas a testes iniciais de triagem para hemoglobinopatias e, posteriormente, foram submetidas à confirmação do perfil eletroforético e cromatográfico por cromatografia líquida de alta performance (HPLC-BIORAD®) e pesquisa de mutações por biologia molecular. A Tabela 1 mostra o resumo dos resultados obtidos na análise das amostras.
A criança apresentava perfil eletroforético de hemoglobina em pH alcalino e ácido compatível com Hb SS mais F (Figura 1). Porém, ao comparar o perfil eletroforético da criança e dos pais, pudemos observar, no perfil de hemoglobina do pai, uma pequena fração, na posição de anidrases carbônicas, sugestiva da variante da globina de cadeia delta.
O cromatograma do filho mostrou o perfil de Hb S (84,9% com tempo de retenção compatível para esta variante) (Figura 2C), Hb A2 (7,9% eluindo com a fração de Hb S acetilada), Hb A0 (2,0%) e Hb F (5,5%). Ao avaliar o perfil dos pais, foi encontrado um perfil de Hb S heterozigoto na mãe, com Hb A0 (51,2%), Hb A2 (4,5% eluindo com Hb S acetilada e, portanto, com valores dentro da faixa normal), Hb F (0,8%) e Hb S (38,4%). O perfil cromatográfico da mãe pode ser visto na figura 2A. O pai apresentava um perfil de Hb AA associado à variante da cadeia delta globina (Hb B2) com os valores percentuais de Hb A0 (84,0%), Hb F (0,2%), Hb A2 (3,3%) e Hb B2 (2,3%); conforme mostrado na figura 2B.
A investigação molecular de mutações hereditárias mostrou a presença de Hb S em heterozigose para a mãe. Para o pai, o valor da Hb A2 adicionado ao valor percentual da variante do alelo delta globina, mostraria um valor para essa fração, acima do limite normal, sugerindo beta talassemia heterozigótica. Assim, investigou-se a presença das quatro mutações mais frequentes da beta talassemia na população brasileira, mostrando heterozigosidade para a mutação IVS1-6 associada à heterozigosidade para a variante de cadeia delta, a Hb B2. O perfil molecular da criança foi confirmado pela presença de Hb S associada à beta talassemia (IVS1-6), justificando assim a clínica mais branda. Foi afastada a herança de alfa talassemia por testes citológicos de rotina e por análise molecular das principais mutações no Brasil.
Tabela 1 – Resultados laboratoriais da família.
| Testes laboratoriais | Pai | Mãe | Filho |
| Morfologia eritrocitária | ++ | N | +++ |
| Resistênica osmótica (NaCl a 0,36%) | + | – | + |
| Eletroforese em pH alcalino | Hb AA+B2 | Hb AS | Hb SF |
| Eletroforese em pH ácido | Hb AA | Hb AS | Hb SF |
| HPLC | Hb AA+B2 | Hb AS | Hb SF |
| Análise molecular | Talassemia b(IVS1-6) + Hb B2 | AS | S/btalassemia (IVS1-6) |
HPLC, Cromatografia líquida de alta performance. Morfologia eritrocitária (++/- microcitose e hipocromia, +++ microcitose, hipocromia e poiquilocitose). Resistência osmótica em solução de NaCl a 0,36% (+ positivo, – negativo).

Figura 1. Perfil de hemoglobina eletroforética em acetato de celulose, pH alcalino, corado com Ponceau. Padrão: Amostra com perfil de hemoglobulina normal (Hb AA). A seta indica a fração referente à Hb B2.
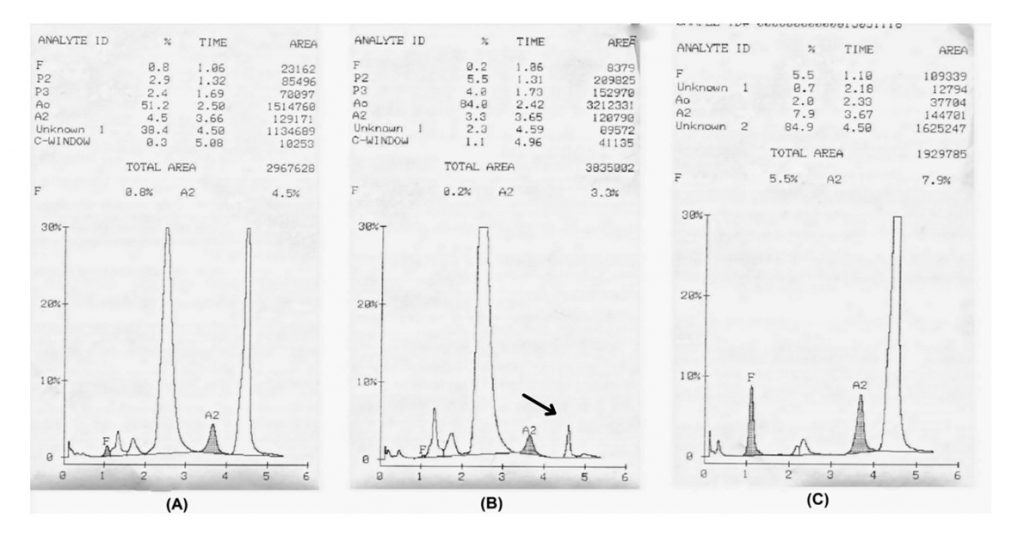
Figura 2. Cromatogramas de cada membro da família, obtidos por equipamentos de HPLC automatizado e kit diagnóstico para variantes e talassemia (BIORAD®).
Em A – perfil cromatográfico e valores percentuais das frações de hemoglobina obtidas na amostra da mãe; Em B – análises da amostra do pai; em C – análise da amostra da criança.
DISCUSSÃO
Considerando que a população brasileira é miscigenada devido ao fluxo migratório de seus grupos populacionais formadores e que os métodos utilizados para identificar variantes de hemoglobina e talassemia na fase neonatal não permitem a identificação completa das interações entre essas diferentes hemoglobinas anormais; faz-se necessário o uso de métodos mais precisos com treinamento técnico específico para identificar as interações menos frequentes da hemoglobina. Assim, este artigo pode contribuir para a elucidação diagnóstica e orientação clínica dos casos que apresentam interações entre hemoglobinas anormais não detectadas no teste de triagem neonatal.
A interação entre Hb S/β tal caracteriza a doença falciforme, bastante comum em nossa população, devido à contribuição dos grupos originais na sua formação, principalmente em regiões onde o fluxo migratório de mediterrâneos e africanos foi intensificado. No diagnóstico laboratorial, a Hb S/β tal apresenta diminuição dos níveis do volume corpuscular médio (VCM) e hemoglobina corpuscular média (HCM), caracterizando uma anemia microcítica hipocrômica, sendo necessários métodos complementares para obtenção do perfil hereditário. A associação de metodologias eletroforéticas e cromatográficas permite a separação de proteínas, com boa resolução, como evidenciado no presente relato de caso.
A migração de hemoglobinas anormais com posições semelhantes pode resultar em um problema no diagnóstico laboratorial, principalmente na associação entre anemia falciforme e talassemia, bem como nos dados hematológicos. Os testes de triagem e o estudo eletroforético convencional não são capazes de distinguir essa interação, sendo necessário um estudo familiar e molecular para confirmação diagnóstica.(2) Outro estudo mostra que os índices hematimétricos da população duplo heterozigótica, como entre Hb S e Hb B2, nem sempre mostram a presença de anemia.(6)
Khalil MSN et al.,(6) 2014 determinaram o espectro mutacional do gene delta globina na população do Reino Unido, e 77% dos casos avaliados foram considerados heterozigotos para a variante de cadeia delta, Hb B2. A hemoglobina foi relatada como a variante da cadeia delta mais comum, observada em quase 1% dos afro-americanos. A origem étnica dos casos foi descrita como afro-caribenha, indígena ou africana. O pico de Hb A2 para esses pacientes teve um tempo de retenção do HPLC entre 4,58 e 4,62 minutos, o que sugere que o HPCL pode ser usado para fazer um pré-diagnóstico da variante anormal, antes da confirmação por técnicas moleculares.
Os resultados do estudo de Khalil MSN et al. 2014,(6) reafirmam a importância de se considerar cuidadosamente o perfil do cromatograma, obtido por HPLC, nos casos com pico de Hb A2 dividido ou com valor de Hb A2 baixo, com índices hematimétricos diminuídos para evitar um diagnóstico incorreto de traço de beta talassemia. Eles também enfatizam a importância de validar os casos suspeitos por análise molecular.
A Hb B2 pode ser confundida ou identificada como Hb A2 e é uma variante silenciosa da cadeia da globina delta com uma frequência de 9,2% em Bantus sul-africanos e entre 1% e 3% em afro-americanos. Na Amazônia brasileira a frequência desta variante correspondeu a 1% dos caucasoides.(7)
Um equívoco bastante comum em testes laboratoriais reside no fato de serem observados os perfis eletroforéticos ou cromatográficos, e não atentar para os valores percentuais e/ou frações minoritárias como a da Hb B2, que interferem nas demais frações e no perfil genético herdado. Valores diminuídos de Hb A2 devem sempre ser objeto de observação cuidadosa, tanto pela herança da alfa talassemia e outras variantes de Hb, como a Hb B2, quanto para perfis de ferro diminuídos, que interferem na produção de todas as hemoglobinas, incluindo aquelas frações que são patognomônicas para diagnósticos mais frequentes.(8-10) A observação cuidadosa de pequenas frações e/ou picos cromatográficos deve ser uma rotina a ser incorporada na prática laboratorial, pois informações importantes podem ser perdidas no diagnóstico.
Ressaltamos também que o olhar atento do clínico quanto ao que esperar da pessoa com hemoglobinopatias e a boa integração com os laboratórios e seus profissionais possibilitam a troca de conhecimentos que facilita a elucidação de casos difíceis e, posteriormente, a orientação adequada para cada caso. O estudo da família é essencial nos casos de heranças complexas dessas proteínas, como a que relatamos neste trabalho.
Os diferentes tipos de associação genética da Hb S, com ênfase na homozigose (Hb SS), interação com beta talassemia, interação com alfa talassemia (Hb SH) e dupla heterozigosidade (Hb SC e Hb SD, principalmente), requerem métodos complementares (eletroforese em pH-ágar ácido, dosagem de Hb fetal, teste de Hb H) e, em particular, o eritrograma, perfil cromatográfico e pesquisa de mutação para estabelecer o diagnóstico real e assim prever o prognóstico para o indivíduo.(11)
Nesse caso específico, a observação clínica do desenvolvimento da criança e o perfil de hemoglobina obtido inicialmente pelos pais já alertavam para uma possível complicação no diagnóstico laboratorial. A criança foi identificada como portadora de anemia falciforme pela presença de Hb SS (SIC) com uma semana de vida (na triagem neonatal). Como não respondeu clinicamente conforme o esperado, novos exames foram realizados, inclusive de seus pais, e os perfis não eram compatíveis para o caso. A investigação aprofundada permitiu o esclarecimento diagnóstico da criança e dos pais, amenizando estresses socioemocionais por diagnósticos equivocados. Os valores percentuais das frações de hemoglobina da criança sugerem a interação da Hb S com a beta talassemia. Porém, o pai foi considerado portador de hemoglobina normal (Hb AA), o que não foi comprovado após a identificação da interação da talassemia β+ (IVS1-6) e variante de cadeia delta (Hb B2), colocando a hemoglobina A2 dentro da normalidade e justificando a discreta microcitose e hipocromia. O perfil mais facilmente identificado foi o da mãe (Hb AS), heterozigota para Hb S.
Com a descrição deste caso, pretendemos chamar atenção para as diferentes nuances do fenótipo da hemoglobina e o quanto a variabilidade genética populacional pode contribuir para a presença de formas interativas.
CONCLUSÃO
Ressaltamos que a identificação das hemoglobinas variantes menos comuns requer uma análise minuciosa com a associação de dados clínicos e diferentes metodologias laboratoriais precisas.
REFERÊNCIAS
- Orlando GM, Naoum PC, Siqueira FAM, Bonini-Domingos CR. Diagnóstico laboratorial de hemoglobinopatias em populações diferenciadas. Rev. Bras. Hematol. Hemoter. 2000; 22(2):111-121.
- Figueiredo MS. The compound state: Hb S/beta-thalassemia. Rev. Bras. Hematol. Hemoter. 2015; 37(3):150-152.
- Melo-Reis PR, Araújo LMM, Dias-Penna KGB, Mesquita MM, Castro FS, Costa SHN. A importância do diagnóstico precoce na prevenção das anemias hereditárias. Rev. Bras. Hematol. Hemoter. 2006; 28(2):149-152.
- Silva LCM, Castro FS. Hemoglobinopatias: Relato de caso familiar. RBAC. 2017;49(3):307-11.
- Vinciguerra M, Passarello M, Leto F, et al. Coinheritance of a rare nucleotide substitution on the β-globin gene and other known mutations in the globin clusters: management in genetic counseling. Hemoglobin. 2016; 40(4):231-235.
- Khalil MSM, Marouf S, Element D, et al. A study of δ-globin gene mutations in the UK population: identification of three novel variants and development of a novel DNA test for Hb A’2. Hemoglobin. 2014; 38(3):201-206.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010; 12:61-76.
- Van Kirk R, Sandhaus LM, Hoyer JD. The detection and diagnosis of hemoglobin A2’ by high-performance liquid chromatography. Am J Clin Pathol. 2005;123(5):657-61.
- Torres FR, Ondei LS, Zamaro PJA, et al. Identificação de HbAB2 em dois caucasianos da região amazônica por procedimentos eletroforéticos e cromatográficos. Rev Bras Hematol Hemoter. 2005;27(2):138-47.
- Naoum PC, Bonini-Domingos CR. Dificuldades no diagnóstico laboratorial das hemoglobinopatias. Rev. Bras. Hematol. Hemoter. [Internet]. 2007: 29(3): 226-228.
- Ferreira N, Zamaro PJA, Bonini-Domingos RR. Interação entre Hb B2 e Hb S. Rev. Bras. Hematol. Hemoter. 2010; 32 (1): 80-82.
Correspondência
Luiz Arthur Calheiros Leite
Avenida Fernandes Lima, 4º andar, Farol
CEP 57050-000 – Maceió – AL – Brasil
Tel.: (55 82) 3432-3230
E-mail: [email protected]