Hemoglobinopatias: relato de caso familiar
Hemoglobinopathies: family case report
Luiza Cristina de Moraes Silva1
Frank Sousa Castro1
1Acadêmica. Pontifícia Universidade Católica de Goiás – PUC Goiás – Goiânia – GO, Brasil.
2Mestre. Pontifícia Universidade Católica de Goiás – PUC Goiás – Goiânia – GO, Brasil.
Instituição: Pontifícia Universidade Católica de Goiás – PUC Goiás – Goiânia – GO, Brasil.
Artigo recebido em 15/02/2017
Artigo aprovado em 20/07/2017
DOI: 10.21877/2448-3877.201700567
Resumo
As hemoglobinopatias, devido à sua alta morbidade e mortalidade, têm sido consideradas um problema de saúde pública no Brasil, onde ainda existem muitos casos de pacientes sem diagnóstico e aconselhamento genético, para que possam então decidir sobre a geração de sua prole. O presente estudo analisou o caso de uma criança com suspeita de anemia falciforme juntamente com seu grupo familiar composto por trinta indivíduos. Os testes realizados foram: eritrograma, eletroforese de hemoglobina em pH alcalino, teste de falcização, dosagem de hemoglobina A2. Indivíduos do grupo familiar com casos inconclusivos foram submetidos a Cromatografia Líquida de Alta Performance. Os resultados obtidos foram: três casos de portadores de traço falciforme, cinco casos de b-talassemia e um indivíduo portador de HbAS/b-talassemia. Os resultados demonstraram a importância do diagnóstico precoce e da triagem populacional para os indivíduos pertencentes a este grupo familiar; assim poderão ser informados, através de aconselhamento genético, sobre os possíveis riscos para a geração futura de sua família, pois trata-se de uma doença que causa anemia hemolítica crônica incurável.
Palavras-chave
Anemia falciforme; Talassemia beta; Hemoglobinopatias; Traço falciforme; Aconselhamento genético
INTRODUção
Hemoglobinopatias são doenças que acometem os genes responsáveis pela síntese das hemoglobinas. O principal exemplo é a anemia falciforme, que é uma hemoglobinopatia genética, hereditária, de alta morbimortalidade.(1) Essa anemia é caracterizada por um mutação pontual (GAG-GTG) nos genes da globina beta da molécula de hemoglobina, acarretando substituição de aminoácido ácido glutâmico por valina na sexta posição, originando a hemoglobina anômala S (HbS).(2)
A hemoglobina S pode se manifestar de três formas: Anemia falciforme, doença falciforme e traço falciforme. A anemia falciforme manifesta-se em indivíduos homozigotos para a hemoglobina S, a doença falciforme em pacientes heterozigóticos em combinação com outras hemoglobinas anormais com grau de gravidade variado: interação da hemoglobina C (Hb SC), associação com talassemia beta (lesão parcial [b+] ou total [b°] do gene b),(3) e o traço falciforme, que se manifesta em interação com hemoglobina normal A. As HbS e C são de origem da raça negra, enquanto que as talassemias são de origem mediterrânea. Porém, a intensa miscigenação no Brasil favorece o aparecimento de associações de hemoglobinopatias em um mesmo indivíduo (HbS/talassemias).(4)
As complicações clínicas podem ser raras para alguns indivíduos que possuem a mutação em apenas um gene (AS), enquanto que, para outros, associados a determinado fatores químicos e ambientais, a sintomatologia pode ser relativamente frequente, conferindo a estes indivíduos uma importância clínica.(5) As características físico-químicas das moléculas da hemoglobina S diferentemente de Hemoglobina A, levam a polimerização, ocasionando a deformação dos eritrócitos, que passam a ser denominados de células em foice.(2)
As alterações laboratoriais encontradas com maior frequência na anemia falciforme são diminuição nos valores séricos de hemoglobina e hematócrito, aumento do número de reticulócitos e diminuição no tempo de vida das hemácias. O diagnóstico laboratorial da anemia falciforme consiste na identificação da HbS por meio dos testes, de eletroforese de hemoglobina em pH alcalino, focalização isoelétrica e ou cromatografia líquida de alta performance (HPLC), que é considerado padrão ouro.(6)
Dentre as complicações clínicas mais frequentes em pacientes portadores da anemia falciforme estão a anemia hemolítica crônica, a vasculopatia cerebral, insuficiência renal, pulmonar e cardíaca, as crises dolorosas agudas osteoarticulares, as lesões crônicas com asplenia funcional, que ocorrem por conta da oclusão da microvasculatura com isquemia tecidual.(2,7)
A b-talassemia em sua forma heterozigota (b-talassemia menor) tem como característica a elevação dos níveis séricos de Hb A2 e/ou Hb Fetal em casos clássicos, e em sua forma mais grave, a b-talassemia maior, caracteriza-se geralmente pela ausência completa da síntese de cadeias beta globínicas e anemia grave.(8,9)
Esse artigo teve como objetivo, avaliar um grupo familiar, e demonstrar a importância do diagnóstico precoce das hemoglobinopatias, tanto em indivíduos em homozigose quanto em heterozigose.
Material e Métodos
Todos os indivíduos do estudo tiveram cadastros realizados e os resultados liberados, no sistema de gestão laboratorial PC Lab Online®. Todas as etapas realizadas no trabalho ocorreram no período de Dezembro de 2015 a Abril de 2016. O trabalho foi realizado com aprovação do Comitê de Ética e Pesquisa da PUC-GO, parecer 235.376 de 20/03/2013.
Grupo familiar
Foi realizado um estudo em um grupo familiar contendo trinta indivíduos, sendo pai e avô da criança não pertencentes à mesma linhagem sanguínea, atendidos no Laboratório Clínico da Pontifícia Universidade Católica de Goiás (LAC-PUC-GO), sob investigação de traço falciforme.
Exames laboratoriais
Todos os indivíduos foram submetidos à avaliação do eritrograma, eletroforese de hemoglobinas em pH alcalino, teste de falcização e dosagem de hemoglobina A2. Para a realização dos exames foram coletados de 4 mL a 5 mL de sangue total, em tubo contendo anticoagulante EDTA (Figura 1).
Eritrograma
Os eritrogramas foram realizados em Contador Hematológico XE-2100D da Sysmex®, que determinou os seguintes parâmetros: hemácias, hemoglobina total, hematócrito, índices hematimétricos (VCM, HCM, CHCM) e o grau de anisocitose (RDW).
Eletroforese de Hemoglobinas em pH alcalino
Para a análise qualitativa das frações de hemoglobina, as moléculas foram separadas por eletroforese de hemoglobina em pH alcalino em fita de acetato de celulose e tampão Tris-glicina (TEB), sendo que o hemolisado foi preparado em uma placa de porcelana com 100 µL de sangue total e 200 µL de saponina a 1%. Em uma cuba eletroforética foram adicionados 50 mL de TEB em cada lado da cuba, com a fita estendida sobre a ponte do aparelho. A leitura dos resultados foi realizada após quarenta minutos, segundo técnica descrita por Naoum PC.(2)
Teste de Falcização
Utilizaram-se 5 µL de sangue total e 10 µL de metabissulfito de sódio a 1% aplicados em uma lâmina, cobertos por uma lamínula vedada com esmalte. Feito isso, a lâmina foi colocada em câmara úmida em repouso por 24 horas e, após, realizada a leitura segundo técnica descrita por Naoum PC.(2)
Dosagem de Hemoglobina A2
A dosagem de Hemoglobina A2 foi realizada pelo método de intercâmbio iônico sem interferência de HbS utilizando o kit comercial da BioSystems®. O hemolisado foi preparado com 50 µL de sangue total e 200 µL de água purificada. Após, o procedimento foi realizado de acordo com as recomendações do kit. Foi feita a leitura em espectrofotômetro da marca Celm®, em comprimento de onda de 415 nm.
Relato de Caso
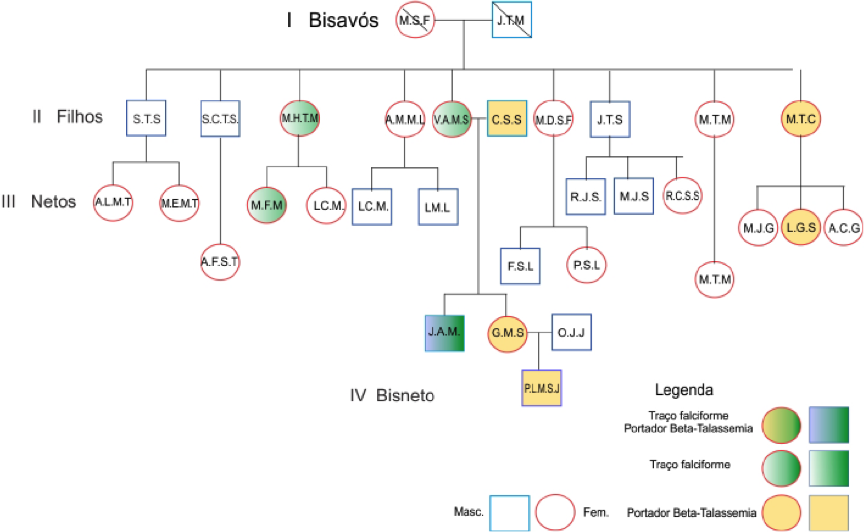
Paciente PLMSJ, sexo masculino, seis meses de idade, foi encaminhado para uma entidade de defesa de direitos, filantrópica e de utilidade pública de sua região após realização do teste do pezinho com suspeita de anemia falciforme. Ao comparecer à unidade, os pais foram informados que o sangue do mesmo ainda estava muito neonato e não poderia ser concluído o diagnóstico. Foram orientados a retornar à unidade quando a criança completasse 1 ano para realizar novos exames. A mãe GMS, e o pai OJJ procuraram o LAC-PUC-GO, para realização de exames e investigação da possível doença. Foi então iniciado um estudo do grupo familiar, onde todos os familiares procuraram o laboratório para a realização dos exames. Os bisavós maternos da criança, já falecidos, possuem dez filhos, dos quais nove compareceram à unidade com seus respectivos descendentes para realização dos exames, não sendo possível obtenção de dados do grupo familiar paterno (Figura 1)
Histórico familiar
A avó da Criança, VMAS, 57 anos, apresentou resultados compatíveis com traço falciforme (eritrograma normal, eletroforese em pH alcalino HbA/S, teste de falcização positivo).
O avô da criança, CSS, 58 anos, apresentou resultado compatível b-talassemia menor (eritrograma: microcitose moderada, hipocromia leve, anisocitose e presença de hemácias em alvo, dosagem de HbA2 – 4,5% [VR= 1,30 a 3,70%]). O tio materno, JAMS, 29 anos, apresentou uma associação de HbS e b-talassemia menor (eritrograma normal, eletroforese em pH alcalino perfil HbA/S, teste de falcização positivo, dosagem de HbA2 – 4,90%).
A tia-avó, MHTM, 48 anos, e uma de suas filhas (LCMS, 22 anos) obtiveram resultados compatíveis com traço falciforme (MHTM: eritrograma normal, eletroforese em pH alcalino perfil HbA/S, teste de falcização positivo, Dosagem de HbA2 – 2,48%. LCMS: eritrograma normal, eletroforese em pH alcalino perfil HbA/S, teste de falcização positivo, dosagem de HbA2 – 3,10%). MTC, tia-avó, 69 anos, e uma de suas filhas (LGS) obtiveram resultados compatíveis com b-talassemia menor (MTC: heritrograma: anemia normocítica e normocrômica, eletroforese em pH alcalino perfil HbAA, teste de falcização negativo, dosagem de HbA2 – 4,64%. LGS: eritrograma normal, eletroforese em pH alcalino perfil HbAA, teste de falcização negativo, dosagem de HbA2 – 4,50%). Os demais indivíduos do grupo familiar apresentaram resultados dentro dos valores de referência esperado (Figura 2)
Histórico da criança
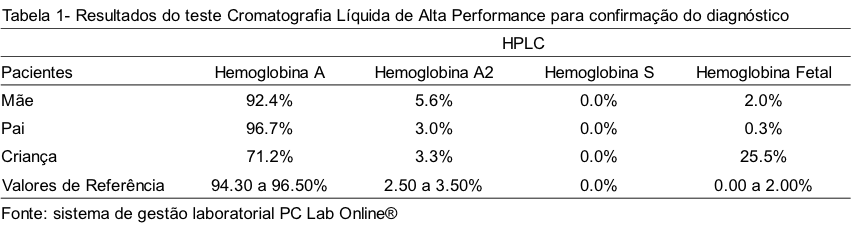
PLMS, no primeiro ano de vida, o eritrograma revelou anemia microcítica e hipocrômica. Ao se observar a lâmina, foi constatada a presença de microcitose moderada, hipocromia leve, anisocitose e hemácias em alvo. A eletroforese em pH alcalino revelou perfil HbA/F, o teste de falcização foi negativo, dosagem de HbA2 – 3,50%. A mãe apresentou no eritrograma alterações de VCM e HCM; em microscopia foi observada microcitose e hipocromia leve. A eletroforese em pH alcalino revelou perfil HbAA, teste de falcização negativo, dosagem de HbA2 – 5,20%. O pai da criança apresentou todos os resultados dentro dos padrões de normalidade. Ao comparecer novamente na unidade, no dia 07/04/2016, após a criança completar um ano, novos exames foram realizados na mãe, na criança e no pai pelo método de Cromatografia Líquida de Alta Performance (HPLC), para elucidação dos resultados inconclusivos, que confirmaram a elevação dos níveis de Hb A2 da mãe e Hb Fetal da criança e os resultados normais do pai (Tabela 1).
Os dados analisados mostraram que a criança, possui b-talassemia menor. Todos os membros do grupo familiar até então não possuíam nenhum tipo de diagnóstico.
Foi possível observar que a mãe e o avô materno são portadores de b-talassemia menor, e a avó materna, portadora de traço falciforme. Ambos não tiveram o aconselhamento genético para decidir sobre a geração de sua prole. Assim, o filho mais novo e tio da criança herdou cada um dos genes dos pais, sendo portador de HbS/b-talassemia menor. Gasparini et al.(4) também observaram em um relato de caso na Argentina a associação entre b-talassemia e hemoglobinopatia S, reforçando a associação entre ambas as alterações, devido à intensa miscigenação mundial.
Guimarães et al.(10) descreveram a importância do aconselhamento genético na resolução de problemas no campo da hereditariedade, ajudando a compreender sobre as características da doença, os possíveis riscos de recorrência em seus familiares, envolvendo todos os aspectos, sendo eles educacionais e reprodutivos, para que eles possam decidir futuramente sobre a sua continuação familiar.
No histórico familiar foram observados dois casos de beta talassemia e três casos de traço falciforme em pacientes isolados.
Segundo o Programa Nacional de Triagem Neonatal (PNTN) de 2008, do Ministério da Saúde, nascem no Brasil 3 mil crianças por ano com Doença Falciforme (DF) e 200 mil com traço falciforme. Devido à prevalência elevada no Brasil, é considerado um problema de saúde pública.(3,5) Como os bisavós da criança são falecidos não foi possível esclarecer a origem da b-talassemia menor ou a origem do traço falciforme.
Conclusões
O aconselhamento genético para portadores heterozigotos do traço falciforme é recomendado pela Organização Mundial da Saúde (OMS), evidenciando o diagnóstico precoce, pois se trata de um problema de saúde pública. O casamento de dois heterozigotos HbA/S ou o casamento de indivíduos HbA/S com heterozigotos portadores de outras hemoglobinopatias, como, por exemplo, a b-talassemia menor, relatada no presente estudo, pode gerar crianças com anemias hemolíticas crônicas e incuráveis. Essas pessoas que apresentam risco de gerar filhos com a doença falciforme têm o direito de serem informadas pelo aconselhamento genético dos possíveis riscos e características da doença, respeitando os aspectos hereditários e demais informações clínicas da doença.
Abstract
Hemoglobinopathies, due to their high morbidity and mortality, have been considered a public health problem in Brazil, where there are still many cases of patients without diagnosis and genetic counseling, so that they can then decide on the generation of their offspring. The present study analyzed the case of a child suspected of having sickle cell anemia together with his family of 30 individuals. The tests performed were erythrogram, hemoglobin electrophoresis at alkaline pH, sickle cell test, hemoglobin A2 dosage. Individuals from the family group With inconclusive cases were submitted to High Performance Liquid Chromatography. The results were: three cases of patients with sickle cell trait, five cases of b-thalassemia and one individual with HbAS/b-thalassemia. The results demonstrated the importance of early diagnosis and population screening for individuals belonging to this family group, thus, they can be informed through genetic counseling about the possible risks to the future generation of their family, since it is a disease that causes incurable chronic hemolytic anemia.
Keywords
Sickle cell anemia; beta-Thalassemia; Hemoglobinopathies; Sickle cell trait; Genetic counseling
REFERÊNCIAS
- Eufemia J, Christine M, Marilyn S, Judith EB, Marsha TLS. Changes in sleep, food intake, and activity levels during acute painful episodes in children with sickle cell disease. J Pediatr Nurs. 2006;21(1):23-34.
- Manual de Diagnóstico e Tratamento de Doenças Falciformes. Brasília: Anvisa, 2002. 142p.
- Pace B, editor. Renaissance of sickle cell disease research in the Genome Era. London: Imperial College Press; 2007.
- Gasparini NP, Agriello EE, Zanella MJ, Jommi MP, Maradei J, Sandoval MJ. Síndrome Drepanocítico. Asociación De Hemoglobina S Y b Talasemia. Medicina B Aires. 2016;76(6):369-72.
- Ministério da Saúde (Brasil), Secretaria de Atenção à Saúde, Departamento de Atenção Especializada. Doença falciforme: Condutas básicas para tratamento. Brasília: Editora do Ministério da Saúde, 2012. 13-15p.
- Steinberg MH. Pathophysiologically based drug treatment of sickle cell disease. Trends Pharmacol Sci. 2006;27(4):204-10.
- Zago M. A anemia falciforme e doenças falciformes. Manual de doenças mais importantes por razões étnicas na população afrodescendente. Brasília: Ministério da Saúde; 2001. p. 13-35.
- Domingos ALB, Granzotto LA, Junior EB, Oliveira TYK, Domingos ACB, Bonini-Domingos CR. Perfil de beta talassemia heterozigota obtido a partir de análise data mining em banco de dados [Carta]. Rev. Bras. Hematol. Hemoter. 2009;32(1):78-9.
- Trigo LAMC, Surita FG, Parpinelli MA, Pereira BG, Fertrin KY, Costa ML. Talassemia beta maior e gestação na adolescência: relato de dois casos. Rev Bras Ginecol Obstet. 2015;37(6):291-6.
- Guimarães CTL, Coelho GO. A importância do aconselhamento genético na anemia falciforme. Cien & Sau Col, 15(Supl. 1):1733-1740, 2010.
Correspondência
Luiza Cristina de Moraes Silva
Pontifícia Universidade Católica de Goiás
Escola de Ciências Médicas, Farmacêuticas e Biomédicas
Avenida Universitária, 1440 – Setor Universitário
74605-010 – Goiânia – GO, Brasil