Diagnóstico hematológico e molecular das neoplasias mieloproliferativas crônicas BCR-ABL negativas
Hematological and molecular diagnosis of BCR-ABL negative chronic myeloproliferative neoplasms
Ana Carolina Menezes Mendonça Valente1
Raquel Tognon Ribeiro2
1 Programa multicêntrico de pós-graduação em Bioquímica e Biologia Molecular, Universidade Federal de Juiz de Fora – Campus Governador Valadares. Governador Valadares, MG, Brasil.
2 Professora Doutora, Departamento de Farmácia, Instituto Ciências da Vida, Universidade Federal de Juiz de Fora – Campus Governador Valadares. Governador Valadares, MG, Brasil.
Recebido em 20/07/2021
Aprovado em 02/03/2022
DOI: 10.21877/2448-3877.202202168
INTRODUÇÃO
As neoplasias mieloproliferativas crônicas (NMPC) configuram um grupo de doenças clonais da célula-tronco hematopoiética, afetando as células da linhagem mieloide. De acordo com a classificação da Organização Mundial da Saúde (OMS) para as neoplasias mieloides publicada em 2016, as NMPC estão divididas em dois grupos: as BCR-ABL positivas, que inclui a leucemia mieloide crônica (LMC), e as BCR-ABL negativas, que inclui a policitemia vera (PV), a trombocitemia essencial (TE) e a mielofibrose primária (MF), classificadas também como cromossomo Philadelphia negativas (Ph-).(1) Ademais, também fazem parte do grupo de NMPC a leucemia neutrofílica crônica (LNC) e a leucemia eosinofílica crônica. A leucemia neutrofílica crônica é caracterizada por leucocitose e hipercelularidade da medula óssea, consistindo predominantemente de células granulocíticas (neutrofilia). Na LNC, existe uma associação com a mutação CSF3R e ausência do cromossomo Philadelphia.(2) A “leucemia eosinofílica crônica – não especificada de outra forma” – é caracterizada pelo aumento de eosinófilos no sangue periférico, pela ausência do cromossomo Philadelphia e por apresentar um aumento de blastos na medula óssea ou no sangue (mas menos de 20% para excluir leucemia aguda como diagnóstico), e a presença de uma anormalidade citogenética clonal e a NMPC inclassificável, a qual inclui neoplasias semelhantes à NMPC que, porém, não podem ser claramente classificadas como os outros subgrupos. A mastocitose fazia parte desse grupo na classificação de 2008, no entanto, em 2016, deixou de ser assim considerada em razão das suas características clínicas e patológicas únicas.(1,2)
O objetivo do presente trabalho é revisar o diagnóstico das principais NMPC (PV, TE e MF), dando enfoque aos achados laboratoriais e ao diagnóstico diferencial. Inicialmente, será apresentada a epidemiologia e fisiopatologia e, posteriormente, será aprofundada a descrição das características clínicas e laboratoriais – achados no hemograma, mielograma, biópsia de medula óssea e exames genéticos.
EPIDEMIOLOGIA
A PV, TE e MF são distúrbios mais frequentes em adultos com 50 a 70 anos com sobrevida mediana estimada de 10 a 15, 15 a 20 e 2 a 5 anos, respectivamente.(3,4) No âmbito global, a mielofibrose, policitemia vera e trombocitemia essencial apresentam taxas de incidência de 0,3 a 1,5; 1,5 a 2,0 e 1,03 a 2,5 por 100.000/ano, respectivamente.(5) Na Europa, um estudo estimou a incidência de PV sendo de 0,68 a 2,6 de 100.000 habitantes, 0,38 a 1,7 para TE e 0,1 a 1,00 para MF e a prevalência estimada foi de 4,96 a 30,00 para PV, 4,00 e 24,00 para TE e 0,51 a 2,7 para MF por 100.000 habitantes.(3) Além da existência de uma grande variação nas estimativas de prevalência e incidência, até o momento não há dados referentes à América do Sul na literatura. Um estudo realizado na Coreia avaliando o período de 2004 a 2013 mostrou um aumento da prevalência das NMPC ao longo dos anos. A TE apresentou faixa de prevalência de 4,1 a 9,0 por 100.000, a PV faixa de 2,8 a 5,4 por 100.000 e a MF faixa de 0,5 a 0,9 por 100.000. Assim, as incidências foram entre 2,0 e 3,0 por 100.000 por ano para TE e 1,0 e 1,5 por 100.000 por ano para PV e a MF variou de 0,3 a 0,5 por 100.000 por ano.(6) A respeito da etnia, brancos apresentaram a maior incidência geral das NMPC, sendo a PV a mais incidente em brancos. Já os leste-asiáticos apresentaram menor incidência, sendo mais frequente o acometimento por TE. Hispânicos e negros apresentaram maior incidência para PV e TE, respectivamente.(6)
Vale ressaltar que em vários países as NMPC são subnotificadas ou classificadas de maneira incorreta, pois a classificação dessas patologias depende de critérios clínicos e laboratoriais que sofreram alterações com a evolução das descobertas a respeito dessas doenças.(7)
Apesar da PV e TE serem doenças com sobrevida longa quando comparadas a outras neoplasias hematológicas, o correto diagnóstico faz-se importante porque esses pacientes podem apresentar complicações e evolução para mielofibrose e leucemia mieloide aguda, conforme será abordado adiante.
FISIOPATOLOGIA
Assim como outras neoplasias, o aparecimento das NMPC está correlacionado com diversos fatores genéticos e ambientais que vão resultar em diferentes fenótipos dessas doenças. Cada uma das NMPC apresenta características mais predominantes quando avalia-se a medula óssea e o sangue periférico, com expectativa de vida e prognóstico diferentes. Porém elas compartilham muitas características relacionadas às disfunções celulares das células hematopoiéticas. Diferentes mutações somáticas adquiridas são responsáveis pela expansão clonal de células-tronco hematopoiética nas NMPC e estão envolvidas em genes relacionados à proliferação e sobrevivência celular. Elas apresentam grande relevância, pois resultam em uma grande variabilidade de manifestações clínicas patológicas.(2,8)
As mutações nos genes JAK2, MPL e CALR são as mutações atualmente conhecidas como responsáveis pelo estabelecimento do fenótipo das NMPC e, por essa razão, são chamadas de “driver mutations”.(9,10) A presença de apenas uma dessas mutações genéticas é o suficiente para o surgimento da patologia, pois todas ativam constitutivamente as mesmas vias a jusante, a via JAK/STAT, a via PI3K/AKT/mTOR, RAS e a via MAPK/ERK, que atuam aumentando a proliferação celular e resistência à apoptose. Dessa forma, culminam em um fenótipo hiperproliferativo e associam-se clinicamente a um risco aumentado de complicações trombóticas, hemorrágicas e evolução à transformação fibrótica ou leucêmica.(8,11,12) Ademais, outras mutações genéticas adquiridas em diversos genes, como TET2, CBL, ASXL1, SH2B3, SF3B1, U2AF1, TP53, IDH2 e EZH2, também podem ser encontradas em pacientes com NMPC, sendo consideradas mutações cooperantes, e podem contribuir para a progressão da doença e transformação leucêmica.(10)
A identificação em 2005 da mutação adquirida JAK2V617F permitiu o melhor entendimento da patogênese dessas doenças. A JAK2V617F é uma mutação somática pontual que consiste na substituição de uma guanina por timina, no éxon 14 do gene JAK2, que acarreta a substituição do aminoácido valina por fenilalanina na posição 617 da proteína. A proteína JAK2 é uma tirosina quinase envolvida na sinalização celular que possibilita a transdução de sinal de diversos receptores de fator de crescimento nas células hematopoiéticas em resposta às citocinas. Por exemplo, a proteína JAK2 atua nas vias disparadas pelos receptores de eritropoetina (EPOR), receptor de trombopoetina (MPL) e receptor de fator estimulante de colônias de granulócitos (G-CSFR).(8,13)
Outra alteração genética importante para as NMPC são as mutações no gene MPL, descrita em 2006. O gene MPL codifica uma proteína que é homóloga a outros membros da superfamília de receptores hematopoiéticos, o receptor de trombopoetina (TPO). Seu ligante, a TPO, atua como regulador da megacariocitopoiese e da formação de plaquetas. Há diversas mutações descritas no gene MPL, mas as principais foram identificadas no éxon 10, a mutação da troca do aminoácido triptofano por leucina W515L ou lisina W515K. Essas mutações acarretam a ativação constitutiva do receptor de TPO e fosforilação das vias de sinalização citadas anteriormente que irão conduzir à proliferação exacerbada de megacariócitos na medula óssea com consequente trombocitose no sangue periférico.(12,13)
Em 2013, foram identificadas em pacientes JAK2V617F negativos mutações somáticas no gene da calreticulina (CALR), que codifica uma proteína que participa da regulação do cálcio intracelular. Acredita-se que a proteína possua atividade relacionada a proliferação, migração, adesão e apoptose celular, através da regulação do cálcio.(14) As mutações somáticas que atingem o gene CALR são do tipo indel, ocorrendo inserção e/ou deleção de pares de bases (pb) no éxon 9 do gene, sendo divididas em dois tipos: a CALRdel52 (deleção de 52pb) ou tipo 1 e CALRins5 (inserção de 5pb) ou tipo 2.(12)
A identificação das mutações JAK2, CALR e MPL é um dos critérios principais para o diagnóstico das NMPC.(1) Um estudo realizado com 162 pacientes, por Porto-Soares,(9) 93,5% dos 61 pacientes com PV apresentaram mutação em JAK2V617F e 1,6% mutação em JAK2 éxon 12. Dos 50 pacientes com TE, 48% apresentaram mutação em JAK2V617F, 22% em CALR e 22% eram triplo negativo. Nos 51 com MF, 61% tinham a mutação JAK2V617F, 25% em CALR e 2% eram triplo negativo; nenhum paciente apresentou mutação em MPL.(9) Geralmente, a mutação JAK2V617F no éxon 14 pode ser encontrada em cerca de 70% das NMPC, está presente em cerca de 95% dos pacientes com PV e em 50% a 60% dos pacientes com TE e MF.(9,10) Nos poucos casos de PV JAK2V617F negativa, ocorrem mutações no éxon 12 de JAK2 em 3% a 5% dos casos de PV. A mutação no éxon 9 do gene da calreticulina (CALR) pode ocorrer em 20% a 25% na TE e 25% a 36% na MF. A mutação no gene do receptor da trombopoetina (MPL) ocorre em cerca de 3% a 20% na TE e 3% a 10% na MF. Aproximadamente 5% a 15% dos pacientes com MF ou TE podem ser “triplo negativos”, ou seja, não apresentar nenhuma das três mutações.(8,10,14) Mais recentemente, um estudo mostrou que pacientes com baixa carga alélica de JAK2V617F também podem apresentar mutações nos genes MPL e CALR, aumentando a complexidade genética.(15)
É possível que ocorram diferentes tipos de combinações de mutações driver e cooperantes em uma célula, e isso implica a grande diversidade fenotípica encontrada em pacientes com NMPC. Além disso, é possível que pacientes que possuem o mesmo perfil genômico apresentem diferentes fenótipos, pois as interações das células não se dão apenas entre as múltiplas mutações que podem acometê-las; a interação com o microambiente também irá refletir no fenótipo.(12) Ainda assim, é importante identificar as mutações driver e cooperantes, bem como suas correlações com o fenótipo e resposta ao tratamento. Por exemplo, o aumento na carga alélica de JAK2V617F está correlacionado com a progressão para MF. Além disso, a presença da mutação em JAK2 aumenta o risco de eventos trombóticos. Dessa forma, a carga do alelo mutante JAK2V617F pode ajudar a determinar o fenótipo da doença.(10) Rumi et al. relataram que a presença da mutação CALR apresenta um risco de trombose menor quando comparado com o da mutação JAK2V617F para os pacientes com PV, por essa razão aparenta oferecer proteção.(13)
CARACTERÍSTICAS CLÍNICAS E LABORATORIAIS DAS NMPC BCR-ABL NEGATIVAS
A PV caracteriza-se laboratorialmente por valores no hemograma que excedem o limite superior do intervalo de referência, apresentando aumento da massa eritrocitária, aumento na concentração de hemoglobina e hematócrito, assim como nas contagens de leucócitos e plaquetas. Clinicamente, pode apresentar-se inicialmente assintomática, mas as alterações hematológicas, inflamatórias e da microvasculatura levam ao aparecimento de sinais e sintomas como fraqueza, cefaleia, eritromelalgia, distúrbios visuais, fadiga, dispneia, perda de peso, prurido, esplenomegalia, sangramento e trombose, sendo a fadiga o sintoma mais comum em PV, apresentando-se em cerca de 85% dos pacientes em um estudo realizado com 1.179 pessoas.(16) Vale ressaltar que ocorrem variações nos sintomas de acordo com o gênero, apesar do gênero não apresentar associação com a classificação de risco. Além disso, alguns sintomas (fadiga, sudorese noturna, febre, dor óssea e prurido) podem estar associados a elevação dos níveis de citocinas em paciente com PV.(16)
Segundo a OMS,(1) o diagnóstico de PV requer a presença dos três critérios principais ou a presença dos dois primeiros critérios principais e o critério secundário.(1) Como critérios principais, considera-se (1) os valores de hemoglobina acima de 16,5g/dL para homens e acima de 16g/dL ou hematócrito acima de 49% para homens e 48% para mulheres, respectivamente); (2) biópsia da medula óssea com hipercelularidade das linhagens eritrocítica, granulocítica e megacariocítica (pan-mielose), com megacariócitos pleomórficos; (3) presença da mutação JAK2V617F ou JAK2 éxon 12. Como critério secundário, considera-se o nível de eritropoetina sérica subnormal.(1) Ressalta-se que os valores de hemoglobina e hematócrito estabelecidos em 2016 foram menores do que os estabelecidos na classificação de 2008, o que teve bastante impacto no diagnóstico dos casos de PV que anteriormente não recebiam esse diagnóstico.(2)
No caso de encontrar apenas dois dos critérios principais é necessário o preenchimento do critério secundário para possibilitar o diagnóstico diferencial de policitemias secundárias. O achado de policitemia (aumento do número de eritrócitos) pode ser secundário ao aumento da produção de eritropoetina (EPO), decorrente, por exemplo, de situações que provocam hipóxia que ocasionam aumento compensatório da EPO, como doenças cardiovasculares, tabagismo, doença renal e alta altitude, ou ainda em decorrência do uso de medicamentos, andrógenos ou existência de tumores.(11) No caso da policitemia secundária, a história clínica e os valores bioquímicos dos pacientes serão diferentes. Para distinguir as condições reativas das condições neoplásicas, além de observar as diferenças morfológicas e o grau de aumento da hematopoese (pan-mielose),(17) realiza-se a dosagem da EPO, já que o nível de eritropoetina é suprimido na policitemia vera. Essa distinção entre PV e policitemia secundária tem impacto sobre o tratamento direcionado à causa do aumento eritrocitário. Por exemplo, na policitemia secundária a realização da flebotomia terapêutica pode ser prejudicial.(11)
O exame de medula óssea na PV mostra aumento da celularidade medular de todas as séries eritroide, granulocítica e megacariocítica (pan-mielose). No entanto, alguns pacientes em fases iniciais de PV e MF podem não apresentar hipercelularidade, se assemelhando mais à TE.(4) Assim, é necessário avaliar também a morfologia dos megacariócitos, conforme detalhado adiante, e também avaliar a presença ou não de displasias para se diferenciar de outras neoplasias mieloides.
Ainda, para o diagnóstico diferencial de distúrbios que cursam com aumento das contagens das células sanguíneas é de grande importância realizar a pesquisa do gene de fusão BCR-ABL para se descartar o diagnóstico de LMC; se negativo, prossegue-se com os testes moleculares para identificar presença de mutações nos genes JAK2, CALR e MPL que auxiliarão na confirmação do diagnóstico e prognóstico.(18)
É possível verificar também algumas alterações bioquímicas na PV, mas são alterações inespecíficas, como hiperuricemia e a hiperuricosúria (em ≥ 80% dos pacientes), hiperbilirrubinemia, VHS diminuída, aumento da fosfatase alcalina de leucócitos (LAP) e lactato desidrogenase sérica (LDH).(17) Vale ressaltar que esses testes não estão entre os critérios diagnósticos para o diagnóstico de PV.
Apesar da expectativa de sobrevida em torno de 10 a 15 anos, os eventos tromboembólicos apresentam grande impacto na qualidade de vida do paciente com PV. Além disso, a doença pode evoluir para leucemia mieloide aguda (transformação em leucêmica) e para mielofibrose pós-PV (secundária).(6,11) A transformação leucêmica ocorre em um tempo médio de 4,6 a 19 anos a partir do diagnóstico inicial de PV,(2) e leva a uma expectativa de sobrevida de apenas 1,5 a 2,5 meses em pacientes não tratados.(16) Alguns podem progredir para um quadro de MF antes de ter transformação leucêmica, sendo que a progressão para MF ocorre em média 8,5 a 20 anos a partir do diagnóstico. Fatores de risco como idade avançada, leucocitose, excesso de citocinas e a presença de mutações exercem influência na progressão da doença.(2,16)
Na TE, 1/3 dos pacientes podem ser assintomáticos e os demais apresentam aumento persistente de plaquetas no sangue periférico > 450×109/L. Os pacientes podem apresentar quadro clínico variável, sintomas como síncope, dor torácica atípica, prurido, perda de peso, distúrbios visuais, fraqueza, parestesias de mãos e pés, esplenomegalia, quadros hemorrágicos e eventos trombóticos.(19)
De acordo com a OMS (2016), o diagnóstico de TE requer o encontro de todos os quatro critérios principais, ou os primeiros três critérios principais mais o critério secundário. Os critérios principais são: (1) número aumentado de plaquetas > 450×109/L; (2) alteração da celularidade na medula óssea, proliferação principalmente da linhagem de megacariócitos com núcleos hiperlobulados; (3) não atender aos critérios da OMS para BCR-ABL1, LMC, PV, MF, síndromes mielodisplásicas ou outras neoplasias mieloides; (4) e a presença de uma das mutações JAK2, CALR, ou MPL. O critério secundário é a presença de um marcador clonal (por exemplo a mutação MPLW515K/L) ou ausência de trombocitose reativa.(1)
O diagnóstico de TE muitas vezes se dá incidentalmente quando pacientes apresentam eventos tromboembolíticos. Para chegar ao diagnóstico de TE tem que ser feita a exclusão de causas reativas comuns de trombocitose. A trombocitose reativa está relacionada com condições infecciosas, inflamatórias, deficiência de ferro, síndromes hereditárias, hemorragia aguda e reações a medicamentos, sendo que todos se caracterizam por serem processos transitórios.(11)
A biópsia de medula óssea na TE mostra principalmente uma proliferação aumentada da linhagem megacariocítica, com megacariócitos grandes com citoplasma abundante, maduros e com núcleos hiperlobulados, aparecendo geralmente agrupados ou dispersos pela medula. Raramente pode haver um aumento em menor grau das fibras de reticulina.(1,20)
O curso clínico da TE caracteriza-se por complicações vasculares que se relacionam à morbimortalidade, estando associadas ao maior risco de trombose venosa profunda e embolia pulmonar.(21) A evolução de TE para MF ou LMA ocorre em cerca de 0,8% a 12,3% e 0,7% a 5,8%, respectivamente.(19)
A MF apresenta como característica laboratorial a presença de anemia com poiquilocitose (principalmente dacriócitos) e leucoeritroblastose (presença de células jovens granulocíticas e eritrocíticas). Na MF pré-fibrótica esses achados podem não ser tão evidentes, podendo o paciente apresentar aumento da contagem de plaquetas e leucócitos, evoluindo posteriormente para leucopenia e trombocitopenia na fase avançada da doença (fibrótica).(22)
Com relação às manifestações clínicas, a MF é caracterizada por sinais e sintomas como fraqueza, cansaço, palpitação e dispneia. É comum o paciente apresentar também esplenomegalia e hepatomegalia resultantes da hematopoiese extramedular. Sintomas de cansaço, mal-estar, perda ponderal, infecções recorrentes e estados febris estão correlacionados com a progressão da doença para o estado fibrótico causado pela diminuição da celularidade da medula.(2)
Os critérios da OMS(1) para o diagnóstico da MF consideram os achados na biópsia de medula óssea, tanto no que diz respeito às atipias dos megacariócitos, como o grau de fibrose, a ausência de critérios para LMC, PV, TE, síndromes mielodisplásicas ou outras neoplasias mieloides, e à presença de mutação JAK2, CALR ou MPL ou presença de outro marcador clonal. O grau de fibrose menor ou igual a 1 caracteriza a fase pré-fibrótica da doença, enquanto grau igual ou maior que 2 caracteriza a fase fibrótica. Além desses critérios maiores, a OMS também estabelece os critérios secundários, sendo necessário que o paciente preencha todos os três critérios principais e pelo menos um critério secundário. Os critérios secundários são: presença de anemia não relacionada a outra comorbidade, leucocitose > 11×109/L esplenomegalia palpável, e LDH acima dos limites normais, sendo que para a fase fibrótica acrescenta ainda um outro critério secundário que é a presença de leucoeritroblastose.(1)
A diferenciação de pré-MF e MF fibrótica é importante por apresentarem quadros distintos nos pacientes e diferentes prognóstico. Os achados no perfil mutacional e morfologia celular da MF na fase pré-fibrótica se assemelham aos encontrados na TE, por isso é necessário diferenciar ambos. A diferença se dá pelas características morfológicas na medula óssea e a clínica da doença. A TE apresenta megacariócitos maiores e maduros e tem melhor sobrevida e menor taxa de progressão leucêmica ou fibrótica, enquanto na fase pré-fibrótica da MF os megacariócitos se apresentam mais agrupados (em clusters) com núcleos hipercrômicos e irregulares.(11,22)
Com o estabelecimento da fibrose na medula óssea, a MF apresenta, na maioria das vezes, um mielograma “seco”, sendo necessária a biópsia da medula. A medula óssea apresenta proliferação de megacariócitos atípicos, com razão núcleo-citoplasma aberrante, núcleos hipercrômicos e irregulares e presença de cluster de megacariócitos. A medula apresenta também fibrose grau 2 ou maior, com fibras de reticulina e colágeno.(1)
Com relação às alterações laboratoriais bioquímicas é possível ocorrer o aumento de LDH, que inclusive é um critério secundário para classificação da MF, e ácido úrico para acima do limite normal superior do intervalo de referência.(23) O LDH foi encontrado aumentado também em outras NMPC, estando correlacionado com o aumento de leucócitos possivelmente por ter propriedade na renovação celular.(24)
Com a progressão da doença, aumentam os blastos no sangue periférico e na medula óssea, sendo que uma porcentagem acima de 20% de blastos indica transformação para leucemia mieloide aguda (LMA). Assim, a avaliação de blastos no sangue periférico auxilia no prognóstico de pacientes com MF, uma vez que a presença de blastos 10% a 19% define uma fase acelerada da doença.(22)
A MF apresenta maior taxa de transformação leucêmica quando comparada às outras NMPC BCR-ABL negativas. A transformação leucêmica causa morte de aproximadamente 20% dos pacientes com MF e fibrose medular.(25) Além da progressão para leucemia, podemos listar como causa de mortalidade desses pacientes a ocorrência de distúrbios cardiovasculares, infecções e sangramentos, esses dois últimos resultantes das citopenias.(22)
Na Figura 1, encontram-se ilustrados os principais achados na PV, TE e MF, de acordo com a presente revisão de literatura.
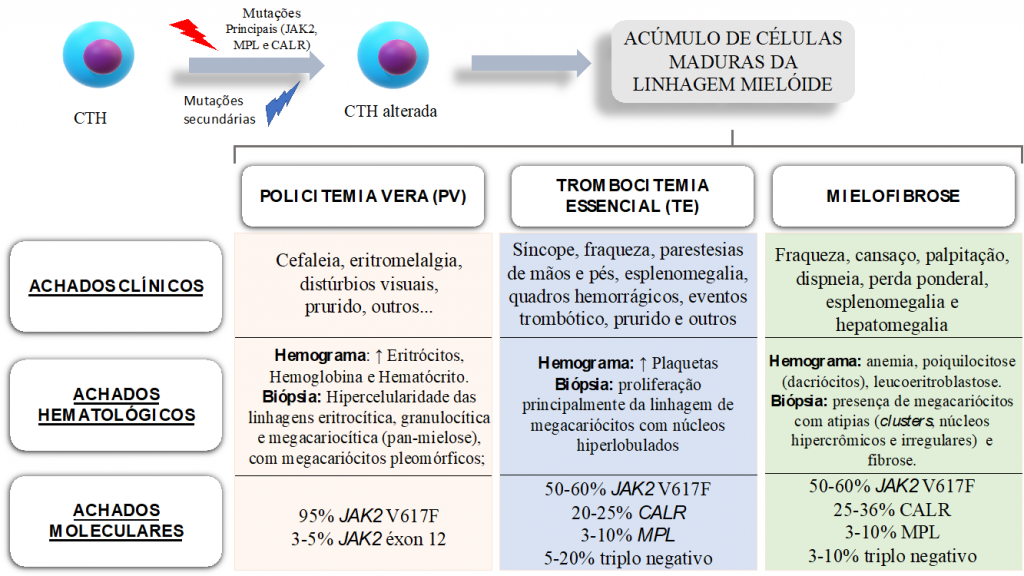
Figura 1. Representação esquemática das alterações moleculares que ocorrem na célula tronco hematopoiética (CTH) e principais achados clínicos, morfológicos e moleculares na policitemia vera (PV), trombocitemia essencial (TE) e mielofibrose (MF).
Fonte: Próprio Autor, com base nas informações apresentadas no artigo.
DIAGNÓSTICO MOLECULAR E AS NEOPLASIAS HEMATOLÓGICAS
O avanço científico e tecnológico permitiu o desenvolvimento de técnicas moleculares que auxiliaram na descoberta de alterações moleculares recorrentes em doenças específicas. Esse conhecimento teve um impacto direto no diagnóstico, prognóstico e definição da terapia para diversas doenças.(26) O desenvolvimento das técnicas de reação em cadeia da polimerase (PCR) e de sequenciamento genético com maior eficiência, maior sensibilidade e menor custo tem permitido que essas técnicas não sejam mais utilizadas somente em pesquisas mas passem a ser cada vez mais utilizadas na prática clínica.(26,27)
A medicina de precisão tem sido utilizada em diversas áreas, mas podemos destacar seu emprego na oncologia. Por exemplo, podemos citar o emprego de pequenos painéis de Sequenciamento de Nova Geração (Next Generation Sequencing, NGS) que vêm sendo utilizados para possibilitar melhor classificação e definição da terapia do câncer de mama.(27)
Para as doenças hematológicas a identificação das diversas alterações moleculares e a utilização desse conhecimento para o diagnóstico, classificação, prognóstico, definição de terapia e monitoramento do tratamento teve uma evolução rápida e expressiva devido à facilidade de obtenção das amostras e estudo das células, quando comparadas aos tumores sólidos.(26)
Em se tratando das neoplasias mieloides, é interessante destacar que, em 2001, a OMS publicou o primeiro documento de classificação das neoplasias hematológicas inserindo critérios genéticos para a classificação da LMA, que passaram posteriormente a serem considerados na classificação de 2008 e de 2016 também para as outras entidades, como as NMPC.(1,28) Como consequência, além do uso da imunofenotipagem por citometria de fluxo e de avaliação das alterações cromossômicas por cariótipo convencional ou molecular (FISH, hibridização in situ fluorescente), o emprego das técnicas de PCR e NGS para detectar as alterações genéticas recorrentes se tornam imprescindíveis para o diagnóstico das neoplasias mieloides.(26)
A leucemia mielóide aguda (LMA) é, portanto, um bom exemplo de como o conhecimento sobre as alterações genéticas impactou na classificação, visto que existe uma categoria específica de classificação para as LMA com as alterações genéticas recorrentes. Para essa leucemia, é necessário pesquisar alterações nos genes que levam à superativação de vias envolvidas na proliferação e diferenciação de células progenitoras hematopoiéticas. As mutações podem ser em genes relacionados a modificações epigenéticas (TET2, IDH1 / IDH2, DNMT3A, ASXL1, KMT2A, EZH2), vias de sinalização (FLT3, KRAS, NRAS, KIT), genes supressores de tumor (TP53, WT1), splicing de RNA (TP3B1), mutação da nucleofosmina (NPM1) e genes que codificam para diferenciação da transcrição (CEBPA, RUNX1).(29)
Também podemos destacar a importância do diagnóstico molecular para outras neoplasias mieloides como a síndrome mielodisplásica (SMD), para a qual podem ser pesquisados genes como SF3B1, ASXL1, DNMT3A, EZH2, RUNX1, SRSF2, TET2, TP53 e U2AF, para a leucemia mielomonocítica crônica (LMMC), para a qual os genes ASXL1, CBL, EZH2, NRAS/KRAS, RUNX1, SETBP1, SRSF2 e TET2 podem ser analisados. Como já mencionado anteriormente, para a leucemia mieloide crônica (LMC), a pesquisa do gene de fusão BCR-ABL é essencial para o diagnóstico e pode ser feita por técnicas como análise do cariótipo para detectar o cromossomo Ph presente em 90% a 95% dos pacientes ou técnicas moleculares como a FISH ou por PCR em tempo real, que também é utilizada para monitoramento terapêutico.(30)
Para as NMPC, como já mencionado, é necessário realizar pesquisa de mutações nos genes JAK2, MPL e CALR. A detecção dessa mutações é bastante útil, pois muitas vezes pode ser difícil diferenciar a LMC, a PV, a TE e a MF em estágios iniciais apenas por critérios morfológicos e até mesmo pode ser difícil de diferenciar de uma leucocitose reativa intensa com desvio à esquerda (benigna).(18)
Na PV, como 95% dos pacientes apresentam a mutação JAK2V617F e os outros 5% apresentam outras mutações no mesmo gene JAK2, o teste para mutação neste gene possui um valor diagnóstico muito grande.(26) Por isso, na classificação OMS essa mutação é incluída como critério maior. Em casos em que o paciente preenche todos os critérios exceto mutação na JAK2, é necessário comprovar que a EPO está normal ou diminuída para se excluir policitemia secundária.(1) Na TE e na MF, cerca de 50% dos pacientes apresentam a mutação na JAK2V617F. Os demais podem apresentar mutações nos genes MPL e CALR, como descrito anteriormente.(10)
Entretanto, é importante notar que várias mutações são comuns entre as neoplasias mieloides e, ainda, que os resultados precisam ser interpretados com cautela devido à chamada CHIP, sigla em inglês para “Hematopoese Clonal de Potencial Indeterminado”, em que o indivíduo já mais idoso apresenta mutações em genes como DNMT3A, TET2, ASXL1, JAK2, TP53, SF3B1, SRSF2 e CBL devido à ocorrência de hematopoiese clonal. No entanto, não significa que ele irá desenvolver obrigatoriamente uma neoplasia mieloide, apesar de terem um risco 10 a 15 vezes maior. Indivíduos saudáveis normalmente apresentam frequência alélica variante (VAF) menor que 30%, visto que VAF > 50% e com mais de uma mutação coexistindo em paciente mais jovem sugerem a presença da doença.(18)
Sistemas de pontuação de prognóstico utilizam a identificação das mutações nos pacientes para prever o prognóstico das NMPC com foco específico em cada fenótipo dessas doenças. Por exemplo, a carga alélica de JAK2V617F e a presença das mutações de alto risco, ASXL1, SRSF2 IDH2, SH2B3, SF3B1, U2AF1 e TP53, têm se mostrado associadas a um pior prognóstico.(10) Pacientes triplo negativos, isto é, negativos para mutações nos genes JAK2, MPL e CALR, apresentam ainda pior prognóstico quando comparados aos pacientes com mutações nesses genes e, nestes casos, é importante investigar mutações nos genes ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2 e SF3B1.(2,29)
CONSIDERAÇÕES FINAIS
Em virtude dos aspectos abordados, a avaliação clínica, morfológica e genética é de extrema importância para o diagnóstico diferencial da PV, TE e MF e os achados nessas avaliações têm impacto direto no prognóstico e tratamento dos pacientes. Além disso, permitem um acompanhamento da evolução dos mesmos. Por sua vez, o avanço científico e tecnológico tem contribuído para a evolução do conhecimento sobre a fisiopatologia, a identificação de mutações e alvos moleculares, refinamento dos critérios diagnósticos e dos sistemas de escore de prognóstico das NMPC. Importante ressaltar que, com o rápido progresso científico na área, torna-se imprescindível que os analistas dos laboratórios de análises clínicas mantenham-se atualizados e busquem se aperfeiçoar nas técnicas moleculares.
REFERÊNCIAS
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Beau MML et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May;127(20):2391-2405. doi: 10.1182/blood-2016-03-643544.
- Barbui T, Thiele J, Gisslinger H, Kvasnicka MH, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in depth discussion. Blood Cancer J. 2018; 8(15):1-11. doi: 10.1038/s41408-018-0054-y.
- Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union Eur J Haematol. 2013 Dec;92(4), 289–97. doi:10.1111/ejh.12256.
- Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017 Feb;129(6):680-92. doi:10.1182/blood-2016-10-695957.
- Harrison CN, Koschmieder S, Foltz L, Guglielmelli P, Flindt T, Koehler M, et al. The impact of myeloproliferative neoplasms (MPNs) on patient quality of life and productivity: results from the international MPN Landmark survey. Ann Hematol. 2017 Aug;96(10):1653–65.doi: 10.1007/s00277-017-3082-y.
- Byun JM, Kim YJ, Youk T, Yang JJ, Jongha Yoo J, Park TS. Real world epidemiology of myeloproliferative neoplasms: a population based study in Korea 2004–2013. Ann Hematol. 2016 Dec; 96(3): 373-81 doi: 10.1007/s00277-016-2902-9
- Titmarsh GJ, Duncombe AS, McMullin MF, O’Rorke M, Mesa R, De Vocht F, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol. 2014 Feb;89(6):581-7. doi:10.1002/ajh.23690.
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017 Feb;129(6):667-79. doi:10.1182/blood-2016-10-695940.
- Porto-Soares MA, de Oliveira RD, Cortopassi GM, Machado-Neto JA, Palma LC, de Figueiredo-Pontes LL, et al. Clinical and molecular profile of a Brazilian cohort of patients with classical BCR-ABL1-negative myeloproliferative neoplasms. Hematol Transfus Cell Ther. 2019;42(3):1-7.doi:10.1016/j.htct.2019.07.008.
- Grabek J, Straube J, Bywater M, Lane SW. (2020). MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells. 2020; 9(8):1-32. doi:10.3390/cells908190.
- Wong WJ, Pozdnyakova O. Myeloproliferative neoplasms: Diagnostic workup of the cythemic patient. Int J Lab Hematol. 2019 Feb;41(Suppl. 1):142–150. doi: 10.1111/ijlh.13005.
- Lee J, Godfreyd AL, Nangaliaa J. Genomic heterogeneity in myeloproliferative neoplasms and applications to clinical practice. Blood reviews. 2020;42.doi: 10.1016/j.blre.2020.100708.
- Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martínez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062–9.doi: 0.1182/blood-2014-05-578435.
- How J, Hobbs GS, Mullally A. Mutant calreticulin in myeloproliferative neoplasms. Blood. 2019 Dec; 134(25): 2242–8.doi:10.1182/blood.2019000622.
- Makarik TV, Abdullaev AO, Nikulina EE, Treglazova SA, Stepanova EE, Subortseva IN, et al. Low JAK2 V617F Allele Burden in Ph-Negative Chronic Myeloproliferative Neoplasms Is Associated with Additional CALR or MPL Gene Mutations. Genes (Basel). 2021 Apr;12(4):559. doi: 10.3390/genes12040559.
- Cuthbert D, Stein BL. Polycythemia vera-associated complications: pathogenesis, clinical manifestations, and effects on outcomes. J Blood Med. 2019;10:359-71.doi:10.2147/jbm.s189922.
- Thiele J, Kvasnicka HM, Muehlhausen K, Walter S, Zankovich R, Diehl V. Policitemia rubra vera versus policitemia secundária. Uma avaliação clínico-patológica de características distintivas em 199 pacientes. Pathol. Res. Pract. 2001;197(2):77-84. doi: 10.1078 / 0344-0338-5710013.
- Zuo Z, Li S, Xu J, You MJ, Khoury JD, Yin CC. Philadelphia-negative myeloproliferative neoplasms: laboratory workup in the era of next-generation sequencing. Curr Hematol Malig Rep. 2019 Aug; 14(5): 376-85.doi:10.1007/s11899-019-00534-8.
- Tefferi A; Pardanani A. Essential Thrombocythemia. N Engl J Med. 2019;381(22):2135- 44.doi: 10.1056 / NEJMcp1816082.
- Fujiwara H. Histological evaluation of myeloproliferative neoplasms. J Clin Exp Hematop. 2018;58(2):45-50. doi: 10.3960/jslrt.18006.
- Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017 Jan;92(1):94-108. doi: 10.1002/ajh.24607.
- Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. 2021;96:145 – 162.doi: 10.1002/ajh.26050.
- Shah S, Mudireddy M, Hanson CA, Ketterling RP, Gangat N, Pardanani A, et al. Marked elevation of serum lactate dehydrogenase in primary myelofibrosis: clinical and prognostic correlates. Blood Cancer J. 2017. 7(12):657 doi:10.1038/s41408-017-0024-9.
- Mudireddy M, Barraco D, Hanson CA, Pardanani A, Gangat N, Tefferi A. The prognostic relevance of serum lactate dehydrogenase and mild bone marrow reticulin fibrosis in essential thrombocythemia. Am J Hematol. 2017 May;92(5):454-459. doi: 10.1002/ajh.24689.
- Masarova L, Bose P, Pemmaraju N, Daver NG, Zhou L, Pierce S, et al. Prognostic value of blasts in peripheral blood in myelofibrosis in the ruxolitinib era. Cancer. 2020 Oct;126(19):4322-31. doi: 10.1002/cncr.33094.
- Ramkissoon LA, Montgomery ND. Applications of next-generation sequencing in hematologic malignancies. Hum Immunol. 2021 Feb 26:S0198-8859(21)00046-X. doi: 10.1016/j.humimm.2021.02.006.
- Zhong Y, Xu F, Wu J, Schubert J, Li MM. Application of Next Generation Sequencing in Laboratory Medicine. Ann Lab Med. 2021 Jan;41(1):25-43. doi: 10.3343/alm.2021.41.1.25.
- Hebeda KM, Fend F. Changed concepts and definitions of myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS) and myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in the updated 2008 WHO classification. J Hematop. 2009 Sep 26;2(4):205-10. doi: 10.1007/s12308-009-0048-6.
- ÖZ Puyan F, Alkan S. The Progress of Next Generation Sequencing in the Assessment of Myeloid Malignancies. Balkan Med J. 2019; 36(2):78-87. doi: 10.4274/balkanmedj.galenos.2018
- Aguilera-Diaz A, Vazquez I, Ariceta B, Mañú A, Blasco-Iturri Z, Palomino-Echeverría S, et al. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PLoS One. 2020 Jan 24;15(1):1-24. doi: 10.1371/journal.pone.0227986.
Correspondência
Raquel Tognon Ribeiro
Universidade Federal de Juiz de Fora – Campus Governador Valadares. Departamento de Farmácia.
Rua São Paulo, 745 – Centro.
Governador Valadares, MG. CEP: 35.010-180
E-mail: [email protected]