Aspectos fisiopatológicos da dislipidemia aterogênica e impactos na homeostasia
Pathophysiological aspects of atherogenic dyslipidemia and impact on homeostasis
Taiane de Macêdo Gondim1
Laise Eduarda Paixão de Moraes1
Italaney Fehlberg2
Vanessa da Silva Brito2
1Especialista em Análises Clínicas pela Escola Bahiana de Medicina e Saúde Pública – Salvador, BA, Brasil.
2Mestre em Imunologia Clínica pela Universidade Federal da Bahia – UFBA – Salvador, BA, Brasil.
3Escola Bahiana de Medicina e Saúde Pública – Salvador, BA, Brasil.
Instituição: Escola Bahiana de Medicina e Saúde Pública – Salvador, BA, Brasil.
Artigo recebido em 23/01/2016
Artigo aprovado em 31/03/2016
DOI: 10.21877/2448-3877.201600462
Resumo
A dislipidemia aterogênica em sinergismo com a existência de transtornos metabólicos como diabetes mellitus tipo 2, síndrome metabólica, hipertensão arterial sistêmica e obesidade, e outros fatores como tabagismo, hábitos alimentares e estresse é reconhecida como um quadro associado a doenças cardiovasculares. Embora o papel da alteração do perfil lipídico neste processo esteja a certo ponto estabelecido, ilustrado por uma elevação das concentrações de CT, triglicerídeos e LDL-C, e da diminuição da HDL-C, os mecanismos pelos quais há a intervenção na hemostasia ainda não estão completamente elucidados, limitando questões clínicas e terapêuticas. No presente trabalho foi realizada uma revisão não sistemática acerca das possíveis alterações hemostáticas associadas à dislipidemia aterogênica descritas atualmente na literatura, o que permitiu uma sumarização dos achados descritos até então.
Palavras-chave
Aterosclerose. Dislipidemias. Homeostasia. Lipoproteínas
INTRODUÇÃO
As dislipidemias são causadas por alterações metabólicas que ocorrem em resposta a distúrbios nas etapas do metabolismo lipídico. Como resultado, o perfil lipídico sérico sofrerá alterações e estas podem incluir aumento do colesterol total (CT), do triglicérides (TG), do colesterol da lipoproteína de baixa densidade (LDL-c) e diminuição do colesterol da lipoproteína de alta densidade (HDL-c).(1) De acordo com a Diretriz Brasileira de Dislipidemias e Prevenção da Aterosclerose (2013), as dislipidemias podem ser classificadas em primárias quando existem bases genéticas, e em secundárias quando associadas a outras doenças, ao uso de medicamentos e/ou ao estilo de vida do indivíduo. A dislipidemia primária pode ainda ser classificada fenotipicamente de acordo com os componentes lipídicos que se apresentam alterados, compreendendo quatro grupos bem definidos: (i) hipercolesterolemia isolada, (ii) hipertrigliceridemia isolada, (iii) hiperlipidemia mista e (iv) diminuição isolada do HDL, com associação ao aumento do LDL e/ou dos TG. Este último perfil se destaca por ilustrar a condição da dislipidemia aterogênica, a qual ainda é geralmente associada a tolerância à glicose prejudicada, resistência à insulina, excesso de peso e/ou gordura corporal, e comorbidades como diabetes mellitus tipo 2 e hipertensão arterial sistêmica. A união destes fatores exerce um efeito sinérgico para o desenvolvimento de doenças cardiovasculares.(2) Esta revisão tem como objetivo discutir possíveis associações entre a fisiopatologia da dislipidemia aterogênica e alterações na homeostasia.
LIPÍDEOS E LIPOPROTEÍNAS
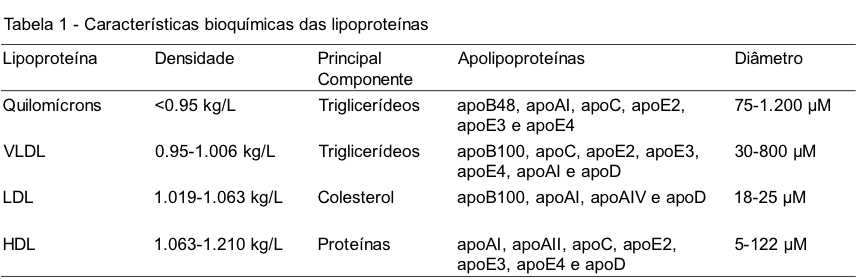
Os lipídios são moléculas orgânicas insolúveis em água resultantes da associação entre ácidos graxos, de cadeias curtas, médias ou longas; e glicerol, estando distribuídos por todos os tecidos do organismo. Entre as principais formas existentes podemos destacar os triglicerídeos e fosfolipídios. Estas formas são as mais abundantes no corpo humano e desempenham funções cruciais para o metabolismo energético. O triglicerídeo é o lipídeo mais comum na alimentação, empregado como fonte de energia para o corpo, enquanto que os fosfolipídios atuam como componente estrutural das membranas celulares. Devido à sua natureza hidrofóbica, os lipídeos são incapazes de circular livremente pelo sangue, sendo necessário um meio de transporte que permita sua distribuição aos órgãos e tecidos. Ácidos graxos de cadeias curta e média podem circular pelo sangue quando ligados à albumina, uma vez que são mais solúveis comparados aos ácidos graxos de cadeia longa. Estes últimos carecem de conversão em triglicerídeos para serem transportados pelas lipoproteínas.(1) As lipoproteínas são moléculas solúveis organizadas de modo a possibilitar o transporte de lipídios entre os tecidos periféricos e a corrente sanguínea.(3) São formadas basicamente por um núcleo hidrofóbico, composto por triglicerídeos e ésteres de colesterol, recobertas por uma membrana anfipática formada por fosfolipídios, colesterol livre e apolipoproteínas (ou apoproteínas), as quais serão discutidas em seguida.(1) Existem quatro classes de lipoproteínas plasmáticas: (i) quilomícrons, (ii) lipoproteína de densidade muito baixa (VLDL), (iii) lipoproteína de densidade baixa (LDL) e (iv) lipoproteína de densidade alta (HDL).(1) Na Tabela 1, é possível observar algumas das características bioquímicas de cada lipoproteína.
QUILOMÍCRONS
Os quilomícrons são as lipoproteínas de maior diâmetro e menor densidade. São responsáveis pelo transporte dos triglicerídeos provenientes da alimentação. A composição consiste em triglicerídeos, colesterol livre, fosfolipídios e uma pequena fração proteica. Os quilomícrons são gerados por células intestinais e secretados na linfa, através da qual alcançam a corrente sanguínea. No sangue, sofrem ação da lipoproteína lipase (LPL), enzima responsável pela degradação dos triglicerídeos em ácidos graxos e glicerol. Os ácidos graxos podem ser oxidados no músculo ou armazenados no tecido adiposo, enquanto que os quilomícrons remanescentes, menores e mais densos, são captados pelo fígado, onde são metabolizados pela ação das enzimas lisossomais dos hepatócitos.(1,3)
LIPOPROTEÍNA DE DENSIDADE MUITO BAIXA (VLDL)
As lipoproteínas de densidade muito baixa (do inglês, Very Low Density Lipoprotein), são sintetizadas no fígado por células parenquimatosas hepáticas e responsáveis pelo transporte do triglicerídeo endógeno para os tecidos periféricos. Logo após a finalização do transporte, estas partículas são hidrolisadas pela ação da LPL e produzem os remanescentes da VLDL, também conhecidos como lipoproteínas de densidade intermediária (IDL). As IDL possuem dois destinos: podem ser captadas e reabsorvidas pelo fígado, ou passar por mais estágios de hidrólise e formar lipoproteínas de baixa densidade (LDL) pela ação da triacilglicerol lipase hepática (HTGL).(1,3)
LIPOPROTEÍNA DE DENSIDADE BAIXA (LDL)
As lipoproteínas de baixa densidade (do inglês, Low Density Lipoprotein) são geradas no estágio final da metabolização das VLDL remanescentes, e representam o principal carreador de colesterol do organismo. São capazes de permanecer por períodos mais longos na corrente sanguínea, sendo por fim captadas pelo fígado via endocitose mediada por receptor, ou pelas células periféricas. É importante destacar que, devido à sua densidade baixa proporcionada pelo alto teor de colesterol, há o favorecimento de sua entrada e alojamento na túnica íntima dos vasos, local no qual sofrem oxidação e podem desencadear o processo de aterogênese pela via scavenger, um evento degenerativo do endotélio vascular.(1,3)
LIPOPROTEÍNA DE DENSIDADE ALTA (HDL)
As lipoproteínas de alta densidade (do inglês High Density Lipoprotein) são sintetizadas no fígado e intestino e responsáveis pelo transporte reverso do colesterol dos tecidos periféricos para o fígado. As HDL nascentes são construídas em parte pelo excesso de fosfolipídios oriundos da hidrólise da VLDL, e pelo colesterol, que retira das células através da ação de uma proteína de membrana que controla de forma limitada a transferência do colesterol livre para a HDL. Após adquirir o colesterol livre, ele é esterificado pela ação da enzima lecitina colesterol acetiltransferase (LCAT), que, por sua vez, será transferido para VLDL, quilomícrons e seus remanescentes em troca de triglicerídeos. Este processo permite que o colesterol retorne à via VLDL-IDL-LDL. Por fim, a HDL, agora rica em triglicerídeos, se liga ao receptor scavenger da membrana dos hepatócitos para transferir o colesterol ao fígado, o que reduz o diâmetro da partícula de HDL e dá origem a uma nova partícula de HDL, a qual participará do próximo ciclo de transporte.(1,3)
APOLIPOPROTEÍNAS
As apolipoproteínas representam a fração proteica das lipoproteínas. Elas se encontram em constante processo de síntese e degradação e são peças fundamentais na regulação do metabolismo lipídico. Exercem funções específicas na regulação do metabolismo lipídico como cofatores enzimáticos, ligantes para os receptores nas células e tecidos pelo organismo, ou através da manutenção estrutural das partículas de lipoproteínas.(3) As apolipoproteínas são divididas em classes (A, B, C, D e E) quanto à composição, o que consequentemente determina função distinta para cada uma das lipoproteínas.(1,3)
APOPROTEÍNA A (apoA)
A apoproteína A (apoA) se apresenta nas isoformas apoA1, apoA2 e apoA4. A apoA1 está presente no sangue principalmente como componente da HDL, nos quilomícrons, e raramente no VLDL, LDL e seus remanescentes.(3) A apo1 é sintetizada no intestino, originalmente como componente dos quilomícrons, e posteriormente transferida para as HDL durante o processo de hidrólise, ou na síntese de novas partículas de HDL.4,5) Como protetor, a apo1 apresenta características antiaterogênica e antioxidante, por ser cofator da enzima LCAT,(3) componente chave para o transporte reverso do colesterol das células para as partículas de HDL e para o fígado.(4) Em indivíduos dislipidêmicos e portadores de diabetes mellitus do tipo 2 com microangiopatias, a apo1 impede a lipotoxicidade e formação da retinopatia através do transporte reverso do colesterol na retina.(4,5) A apo2, sintetizada no fígado, é a segunda proteína mais abundante na HDL.(3) Funcionalmente, a apo2 modula diferentes etapas do metabolismo da HDL, podendo gerar a inibição da atividade da LCAT e da proteína de transferência de colesterol esterificado (CETP), e aumento da atividade lipase hepática, o que contribui para o transporte reverso do colesterol. Por outro lado, a apo2 pode inibir a absorção hepática do colesterol presente na HDL, impactando negativamente no transporte reverso do colesterol. Os efeitos da apo2 na aterogênese, embora controversos, não se revelam muito determinantes para o metabolismo lipídico.(1) A apo4, assim como é apo1, é sintetizada quase que exclusivamente pelo fígado e intestino e tem se mostrado um potente ativador da LCAT.(3)
APOPROTEÍNA B (apoB)
Responsável pelo transporte do colesterol para as células periféricas, a apolipoproteína B (apoB) está presente nos quilomícrons e nas partículas de VLDL, VLDLs remanescentes e LDL. Duas isoformas são relatadas: apoB48 apoB100. A apoB48 é encontrada nas partículas de quilomícrons e remanescentes – de alto potencial aterogênico – e a apoB100 é um componente obrigatório das VLDL, VLDL remanescentes e LDL.(3,5) O metabolismo da apoB48 e da apoB100 são completamente distintos. Enquanto a apoB48 nos quilomícrons é rapidamente metabolizada e absorvida pelo fígado, a apoB100 é inicialmente secretada em partículas da VLDL que, pela ação da lipase, é convertida em IDL e posteriormente em LDL, a qual é metabolizada de forma lenta.(3) A presença da apoB100 no LDL é essencial para facilitar a entrada do colesterol nas células através da sua ligação aos receptores celulares.(4) Esta ligação ao receptor está intrinsicamente relacionada com o acúmulo do colesterol nas artérias, fator desencadeante para o processo aterogênico.(3,6)
APOPROTEÍNA C (apoC)
A apolipoproteína C (apoC) está presente na superfície dos quilomícrons, VLDL e HDL. Apresenta-se nas isoformas apoC1, apoC2, apoC3 e apoC4. Embora exibam funcionalidades metabólicas distantes, todas as apoC compartilham a propriedade de redistribuir componentes entre as lipoproteínas(3) através da ativação da LCAT.(6) A apoC1 inibe a captura de VLDL pelo fígado, além de auxiliar no processo de esterificação do colesterol, o que possivelmente confere à apoC1 uma participação na remodelação da HDL.(3,6) A apoC2 ativa a lipase lipoproteica (LPL), responsável pela hidrólise de partículas ricas em triglicerídeos,(3,6) que, em contrapartida, é inibida pela apoC3, a qual modula a absorção das partículas ricas em triglicerídeos pelos receptores hepáticos. A apoC4 está envolvida na regulação da absorção dos lipídeos.(6)
APOLIPOPROTEÍNA D (apoD)
Presente nas lipoproteínas VLDL, LDL e, em maiores quantidades, na HDL, a apoproteína D (apoD) é pouco relatada devido à sua pequena expressividade. Embora a HDL seja considerada uma molécula antiaterogênica, recentemente foi estabelecido que sua expressão aberrante tem associação com alterações no metabolismo lipídico e na deposição e acúmulo das placas de ateroma no processo aterogênico. Ademais, estudos apontam um possível envolvimento da apoD com a ativação da LCAT e em processos que favorecem o amadurecimento da HDL.(7)
APOPROTEÍNA E (apoE)
Constituintes dos quilomícrons e remanescentes, VLDL e HDL,(3) a apoproteína E (apoE), está empregada na regulação dos níveis plasmáticos dos lipídeos, o que impacta sumariamente antiaterogênese. Ela promove a captação eficiente das lipoproteínas na circulação, desempenha papel no transporte reverso do colesterol, inibe a agregação plaquetária e modula a função imune.(8) As isoformas desta apoproteína são: apoE2, apoE3 e apoE4.(3,9) A apoE3 é associada com a funcionalidade ideal do metabolismo lipídico. Isto em grande parte devido à sua conformação flexível, que permite uma melhor interação com lipídeos de diferentes densidades e tamanhos,(10) enquanto que a apoE2 e apoE4 estão associadas à dislipidemia e risco acentuado para doenças cardiovasculares.(11) É importante ressaltar que a apoE3 é crucial para o correto funcionamento do metabolismo lipídico quando em níveis ideais, determinado pela atuação na endocitose das partículas remanescentes do metabolismo do quilomícron e pela promoção da produção de VLDL pela estimulação intracelular nos hepatócitos. Uma superexpressão ou acúmulo da apoE3 estimula a produção de VLDL, aumentando consequentemente as concentrações de triglicerídeos desta partícula e os níveis de LDL circulante no sangue.(10)
DISLIPIDEMIAS
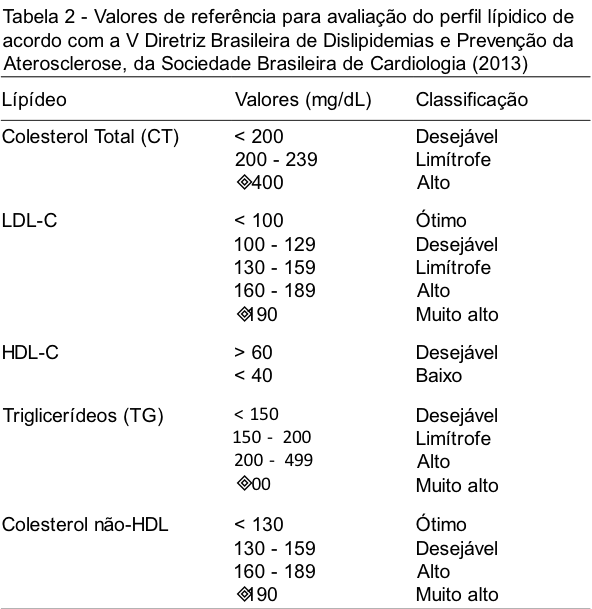
Dislipidemia é o termo utilizado para designar os níveis alterados – na maioria das vezes aumentados – de lipídeos e/ou lipoproteínas por meio da alteração em alguma ou algumas das fases do metabolismo lipídico. Os valores de referência para o perfil lipídico são apresentados na Tabela 2.
Etiologicamente as dislipidemias são classificadas em (i) primárias, quando estão relacionadas a fatores genéticos ou não tem causa aparente, e (ii) secundárias, quando relacionadas a outras doenças, uso de medicamentos ou estilo de vida do paciente.(2) As dislipidemias primárias podem ainda ser classificadas de acordo com suas características genotípicas e fenotípicas. Relacionado ao genótipo, estas podem obter a subclassificação em monogênicas (uma mutação envolvida) e poligênicas (múltiplas mutações envolvidas). A respeito do fenótipo, subclassificadas (classificação laboratorial) ao utilizar os marcadores bioquímicos Colesterol Total (CT), HDL-C, LDL-C e Triglicérides (TG), e divididas em quatro perfis distintos: (i) hipercolesterolemia isolada, (ii) hipertrigliceridemia isolada, (iii) hiperlipidemia mista e (iv) diminuição isolada dos níveis de HDL, com associação do aumento dos níveis de LDL e/ou dos níveis de TG.(2) As dislipidemias secundárias se apresentam associadas a algum transtorno metabólico de base – diabetes mellitus tipo 2,(12) hipertensão arterial sistêmica,(13,14) síndrome metabólica(15,16) e obesidade(17) – à terapia antirretroviral altamente ativa (HAART) de pacientes HIV/AIDS(18) e a fatores ambientais como tabagismo, hábitos alimentares e estresse.(5,19) A compilação de fatores externos e internos exerce um efeito sinergístico para o desenvolvimento de doenças cardiovasculares, agravamento de distúrbios metabólicos ou ainda iniciar o processo de formação de placas de ateroma nos vasos sanguíneos, consequência comum do quadro de dislipidemia aterogênica.(2) O perfil fenotípico da dislipidemia aterogênica é bastante singular e facilmente reconhecido, sendo ilustrado por uma elevação das concentrações de CT, em conjunto com aumento dos níveis de triglicerídeos e LDL, e da diminuição dos níveis de HDL.(17,20,21) A dislipidemia aterogênica muitas vezes não tem um curso favorável devido ao seu diagnóstico tardio e início assintomático.(2) Entende-se por aterogênico todo processo capaz de produzir alterações degenerativas nas paredes das artérias. O endotélio arterial em estado saudável repele as células circulantes no sangue e é fortemente antitrombótico. Na aterogênese, uma agressão endotelial permite o depósito de lipídeos na camada íntima do vaso, o que promove a formação de placas de ateroma, acompanhadas por uma reação inflamatória local. Em longo prazo, a presença destas placas de ateroma ocasiona a oclusão completa da luz arterial, e como consequência pode haver infarto agudo do miocárdio ou um acidente vascular cerebral (AVC), dependendo do local de acúmulo das placas.(1)
DISLIPIDEMIA ATEROGÊNICA
Em condições fisiológicas normais, o endotélio vascular promove alterações funcionais adaptativas para a manutenção da hemodinâmica, por meio da liberação de substâncias com propriedades antiaterogênicas, sendo o óxido nítrico (NO) a principal delas. O NO, em condições ideais, limita o recrutamento vascular de leucócitos, impedindo sua adesão à parede vascular e inibindo a agregação plaquetária, o que evita a formação de trombos. A perda da atividade biológica do NO é denominada disfunção endotelial.(13)
Inicialmente, o dano é mais funcional do que estrutural. O endotélio perde sua habilidade de repelir as células inflamatórias circulantes no sangue e passa a permitir a sua adesão na parede vascular, o que o torna permeável às lipoproteínas, culminando, a longo prazo, em dano estrutural.(3) O aumento da permeabilidade endotelial favorece a entrada da LDL-C para a região íntima vascular, onde não são capazes de serem absorvidas, o que favorece seu acúmulo e oxidação.(19) O acúmulo de LDL, unido à proliferação de células para a luz do vaso arterial, tem sido apontado como um fator desencadeante para aterogênese, por meio da promoção de reação inflamatória local exacerbada.(13,14,22) Unido a isto, o aumento dos níveis de LDL-C gera um maior consumo de NO com liberação de radicais livres, os quais, por sua vez, promovem a oxidação da LDL-C acumulada, formando a LDL oxidada (LDLox), partículas que possuem potencial aterogênico acentuado e elevada citoxicidade. A citotoxidade da LDLox agrava a disfunção endotelial, gerando a expressão de quimiocinas pelas células lesionadas do endotélio, favorecendo o recrutamento de neutrófilos para o interior do vaso sanguíneo.(14,22) Paralelamente, a LDLox estimula também a migração de monócitos e sua diferenciação em macrófagos, os quais endocitam as partículas oxidadas através dos receptores scavengers e promovem a formação das células espumosas – componentes essenciais da placa aterosclerótica.(13,14,22) A endocitose das LDLox exerce papel ambíguo na fisiopatologia da doença: enquanto possui teor protetor por conta da remoção das partículas oxidadas, o mesmo processo também induz a produção dos mesmo radicais livres responsáveis por agravar a lesão endotelial, consequentemente promovendo a expressão de mais quimiocinas, as quais, por sua vez, irão recrutar novos monócitos e estimular a projeção das células musculares lisas para a luz do vaso, estabelecendo o dano estrutural. Isto desencadeia um processo sistêmico que envolve proteínas da coagulação e promove a evolução da lesão aterosclerótica por meio do perpetuamento do processo inflamatório.(14,22)
Na Figura 1 é possível visualizar a patogênese da aterosclerose.
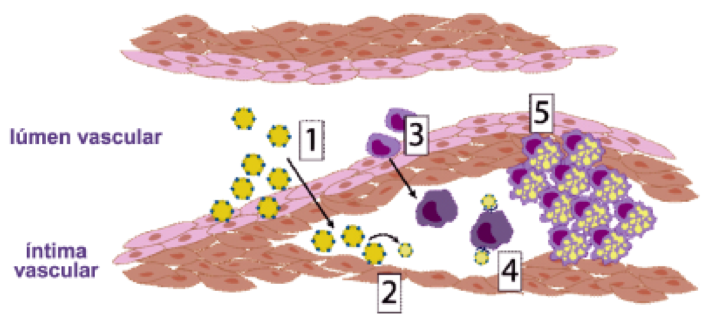
Figura 1. Resumo do processo aterogênico: (1) o efluxo de macromoléculas LDL para a camada íntima vascular (2) gera o acúmulo e oxidação destas, originando a formação das LDLox, agravando a disfunção endotelial devido à sua citotoxicidade. (3) A LDLox estimula a migração de monócitos e sua diferenciação em macrófagos, (4) os quais fagocitam as LDLox através dos receptores scavengers. (5) Após fagocitarem as LDLox, os macrófagos se transformam em células espumosas que promovem, por meio de processo inflamatório, a projeção das células musculares lisas para o lúmen vascular.
DISLIPIDEMIA ATEROGÊNICA NA HOMEOSTASIA
A cascata da coagulação é responsável pela manutenção da fluidez sanguínea pela formação de coágulos de fibrina em casos de lesão vascular. Entre as alterações nas proteínas envolvidas no processo de coagulação são descritas elevações séricas nos níveis do inibidor do ativador do plasminogênio tipo 1 (PAI-1), o inibidor da fibrinólise ativado pela trombina (TAFI) , do fator III (tissular) e VII da coagulação.(24,25) A baixa concentração dos níveis de HDL associada aos altos níveis de LDLox é ineficaz na inibição do fator de agregação plaquetária. Somado a isto, a hipertrigliceridemia característica da dislipidemia aterogênica estimula os fatores III e VII da coagulação, responsáveis por iniciar a via extrínseca da cascata da coagulação, o que favorece a formação de trombos no lúmen dos vasos sanguíneos.(24) A presença da LDLox das placas de ateroma aumentam a expressão do fator III pelas células do músculo liso do endotélio vascular, o qual atua como importante promotor da migração e proliferação de células do músculo liso para o lúmen do vaso sanguíneo e da angiogênese.(23) Em paralelo à indução dos fatores III e VII, o acúmulo de lipídeos eleva os níveis dos fatores da coagulação PAI-1 e TAFI, promovendo a inibição do processo fibrinolítico responsável por dissolver e retirar da circulação coágulos que possam obstruir o fluxo sanguíneo. Deste modo, tanto a fisiopatologia inerente à dislipidemia aterogênica quanto os seus reflexos nos mecanismos da coagulação favorecem um ciclo infindável para a oclusão da luz do vaso sanguíneo.(26)
CONCLUSÃO
O curso fisiopatológico da dislipidemia aterogênica favorece a evolução da aterosclerose e contribui para o estabelecimento de lesões degenerativas crônicas graves. Muitos aspectos relacionados às alterações da homeostasia na presença de hipercolesterolemia e/ou síndrome metabólica são relatados em estudos, no entanto no que se refere especificamente à dislipidemia aterogênica, evidencia-se a necessidade de mais pesquisas, sobretudo epidemiológicas, para traçar o comportamento fenotípico da doença em populações distintas. Adicionalmente, estudos de casos que relatem o impacto positivo ocasionado pela avaliação clínica e prognóstico dos indivíduos, com apresentações de algumas destas alterações discorridas, poderia diminuir substancialmente as chances do estabelecimento de comorbidades mais graves. Em um aspecto mais amplo, e contextualizando com o Brasil, poderia ainda impactar nos gastos voltados para a saúde pública.
Abstract
The atherogenic dyslipidemia in synergy with metabolic disorders such as diabetes mellitus type 2, metabolic syndrome, hypertension and obesity, added to other factors such as smoking, diet and stress is associated with cardiovascular diseases. Although the impact of changes in lipid metabolism in this process is established by a high concentrations of CT, triglycerides and LDL-C, and decreased HDL-C, the effects of these changes in the homeostasis have not been fully elucidated, the effects of changes in the homeostasis have not been fully elucidated, limiting clinical and therapeutic improvements. In the present work a non-systematic review about the possible changes associated with atherogenic dyslipidemia currently described in the literature, which allowed a summarization of the findings described so far. In the present study, a non-systematic review of possible changes in the homeostasis associated with atherogenic dyslipidemia currently described in the literature was made, which allowed a summarization of the mechanisms described so far.
Keywords
Atherosclerosis; Dyslipidemias; Homeostasis; Lipoproteins.
REFERÊNCIAS
- Baynes JW, Dominiczak MH. Bioquímica Médica. 3rd ed. Elsevier; 2011. 680 p.
- SBC. V Diretriz Brasileira de Dislipidemias e Prevenção da Aterosclerose. In: Arquivos Brasileiros de Cardiologia. Sociedade Brasileira de Cardiologia; 2013. p. 01-22.
- Mahley RW, Innerarity TL, Rall SC, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. 1984 Jun;25(12):1277-94.
- Srinivasan SR, Berenson GS. Serum apolipoproteins A-I and B as markers of coronary artery disease risk in early life: the Bogalusa Heart Study. Clin Chem. 1995 Jan;41(1):159-64.
- Lima LM, Carvalho MD, Sousa MO. Apo B/apo A-I ratio and cardiovascular risk prediction. Arq Bras Cardiol. 2007 Jun;88(6):e187-90. [Article in English, Portuguese].
- Forti N, Diament J. Apolipoprotein B and A-I: cardiovascular risk factor? Rev Assoc Med Bras (1992). 2007 May-Jun;53(3):276-82. [Article in Portuguese].
- Perdomo G, Henry Dong H. Apolipoprotein D in lipid metabolism and its functional implication in atherosclerosis and aging. Aging (Albany NY). 2009 Jan;1(1):17-27.
- Davignon J, Cohn JS, Mabile L, Bernier L. Apolipoprotein E and atherosclerosis: insight from animal and human studies. Clin Chim Acta. 1999 Aug;286(1-2):115-43.
- Phillips MC. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life. 2014 Sep;66(9):616-23
- Huang Y, Liu XQ, Rall SC, Taylor JM, Von Eckardstein A, Assmann G, et al. Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. J Biol Chem. 1998 Oct 9;273(41): 26388-93.
- Davignon J, Bouthillier D, Nestruck AC, Sing CF. Apolipoprotein E polymorphism and atherosclerosis: insight from a study in octogenarians. Trans Am Clin Climatol Assoc. 1988;99:100-10.
- Burns SF, Lee S, Bacha F, Tfayli H, Hannon TS, Arslanian SA. Pre-diabetes in overweight youth and early atherogenic risk. Metabolism. 2014 Dec;63(12):1528-35.
- Bahia L, Guilherme L, Aguiar K, Villela NR, Bottino D, Bouskela E. Endotélio e aterosclerose. Rev da SOCERJ. 2004;17:26-32.
- Corrêa-Camacho C. Aterosclerose, uma resposta inflamatória. Arq Ciências da Saúde. 2007;14(1):41-8.
- Lottenberg SA, Glezer A, Turatti LA. Metabolic syndrome: identifying the risk factors. J Pediatr (Rio J). 2007 Nov;83(5 Suppl): S204-8.
- Faria ER, Faria FR, Franceschini Sdo C, Peluzio Mdo C, Sant Ana LF, Novaes JF, et al. Insulin resistance and components of metabolic syndrome, analysis by gender and stage of adolescence. Arq Bras Endocrinol Metabol. 2014;58(6):610-8.
- Pires A, Martins P, Pereira AM, Silva PV, Marinho J, Marques M, et al. Insulin Resistance, Dyslipidemia and Cardiovascular Changes in a Group of Obese Children. Arq Bras Endocrinol Metabol. 2014 Aug;58(6):610-8. [Article in Portuguese].
- Nsagha DS, Assob JCN, Njunda AL, Tanue EA, Kibu OD, Ayima CW, et al. Risk Factors of Cardiovascular Diseases in HIV/AIDS Patients on HAART. Open AIDS J [Internet]. 2015 Oct 20;9(1):51-9. Available from: http://benthamopen.com/ABSTRACT/TOAIDJ-9-51.
- Jorge PA. Endothelium, lipids and atherosclerosis. Arq Bras Cardiol. 1997 Feb;68(2):129-34. [Article in Portuguese].
- Núñez-Cortés JM, Pedro-Botet Montoya J, Pintó Sala X. Riesgo vascular residual: recomendaciones de la Iniciativa Española para la Reducción del Riesgo Residual. Med Clin (Barc). 2010;135 (4):165-71.
- Carr MC, Brunzell JD. Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab. 2004 Jun;89(6):2601-7.
- Nakajima K, Nakano T, Tanaka A. The oxidative modification hypothesis of atherosclerosis: the comparison of atherogenic effects on oxidized LDL and remnant lipoproteins in plasma. Clin Chim Acta. 2006 May;367(1-2):36-47.
- Brand A, Griffiths DJ, Herve C, Mallon E, Venables PJ. Human retrovirus-5 in rheumatic disease. J Autoimmun [Internet]. 1999 Aug;13(1):149-54. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10441180
- Okopien B, Krysiak R, Herman ZS. Effects of short-term fenofibrate treatment on circulating markers of inflammation and hemostasis in patients with impaired glucose tolerance. J Clin Endocrinol Metab. 2006 May;91(5):1770-8.
- Ríos MS, Caro JF, Carraro R, Fuentes JAG. 22. Alterations in Thrombosis and Fibrinolysis in the Metabolic Syndrome. In: The Metabolic Syndrome at the Beginning of the XXI Century: A Genetic and Molecular Approach. Elsevier; 2005. p. 479.
- Lima LM, Carvalho MG, Sabino AP, Sousa MO. Lipoproteína A e inibição da fibrinólise na doença arterial coronariana. Rev Bras Hematol Hemoter. 2006;28(1):53-9.
Correspondência
Taiane de Macêdo Gondim
Rua Airosa Galvão – 7/302 – Barra
40140180 – Salvador, BA